结构优化与频率计算
结构优化的目的是找到体系势能面的极小点。能找到哪个极小点取决于输入文件中提供的初始结构,离哪个极小点越近,一般越容易收敛到哪个极小点。
结构优化在数学上等价于寻找多元函数极值问题:
结构优化常用算法如下:
最速下降法(Steepest descent):最速下降法就是沿着负梯度的方向进行线搜索,对于远离极小点的结构,最速下降法优化效率非常高,但临近极小点时收敛慢,容易震荡。
共轭梯度法(Conjugate gradient):共轭梯度法是最速下降法的改良,每步优化方向与前一步的优化方向相组合,能一定程度缓解震荡问题。
牛顿法(Newton method):牛顿法的思路是将函数相对于当前位置进行泰勒级数展开。牛顿法收敛很快,对于二次函数一步就可以走到极小点。但是牛顿法需要求解Hessian矩阵,计算非常昂贵,一般几何优化中使用准牛顿法。
准牛顿法(Quasi-Newton method):准牛顿法通过近似方法构建 Hessian矩阵,当前步的Hessian矩阵基于当前步的受力和上一步的Hessian矩阵来得到。具体做法有多种,最常用的是BFGS法,此外还有DFP、MS、PSB等。由于准牛顿法的 Hessian是近似构建的,所以每一步优化的准确度低于牛顿法,达到收敛所需步数较牛顿法更多。但由于每一步耗时大为降低,所以优化总耗时还是显著减少了。
BDF的结构优化是由BDFOPT模块来实现的,支持基于牛顿法和准牛顿法来进行极小值点结构和过渡态结构的优化,且支持限制性结构优化等。以下对BDFOPT模块的输入文件格式举例进行介绍,并对输出文件进行解读。
基态结构优化:一氯甲烷( \(\ce{CH3Cl}\) )在B3LYP/def2-SV(P)水平下的结构优化
$compass
title
CH3Cl geomopt
basis
def2-SV(P)
geometry
C 2.67184328 0.03549756 -3.40353093
H 2.05038141 -0.21545378 -2.56943947
H 2.80438882 1.09651909 -3.44309081
H 3.62454948 -0.43911916 -3.29403269
Cl 1.90897396 -0.51627638 -4.89053325
end geometry
$end
$bdfopt
solver
1
$end
$xuanyuan
$end
$scf
rks
dft
b3lyp
$end
$resp
geom
$end
其中RESP模块负责计算DFT梯度。与其他大部分任务不同,在结构优化任务中,程序并不是按顺序、单次、线性地调用各模块,而是会反复多次调用各模块。具体的调用顺序为:
运行COMPASS,读取分子结构等信息;
运行BDFOPT,对结构优化所需的中间量进行初始化;
BDFOPT启动一个独立的BDF进程,用来计算当前结构下的能量和梯度,该进程只执行COMPASS、XUANYUAN、SCF、RESP各模块,而跳过BDFOPT。也即,大部分时候用户会发现有两个BDF进程在彼此独立地运行,其中一个为BDFOPT所属的进程,处于等待状态,而另一个进程则在进行能量和梯度的计算。为避免输出文件过于杂乱,后一个进程的输出会被自动重定向到后缀为
.out.tmp的文件,从而与BDFOPT模块的输出(一般会被用户重定向到.out文件)分开;待后一个进程结束时,BDFOPT汇总当前结构的能量和梯度信息,并据此调整分子结构,以期降低体系能量;
BDFOPT根据当前结构的梯度以及当前几何结构步长(geometry step)的大小,判断结构是否收敛,如收敛,或结构优化达到最大迭代次数,则程序结束;如不收敛,则跳至第3步。
因此, .out 文件只包含COMPASS和BDFOPT模块的输出,可以用来监测结构优化的进程,但不包含SCF迭代、布居分析等信息,后者需要在 .out.tmp 文件中查看。
以上述 \(\ce{CH3Cl}\) 的结构优化任务为例,可以看到 .out 文件里BDFOPT模块的输出:
Geometry Optimization step : 1
Single Point SCF for geometry optimization, also get force.
### [bdf_single_point] ### nstate= 1
Allow rotation to standard orientation.
BDFOPT run - details of gradient calculations will be written
into .out.tmp file.
...
### JOB TYPE = SCF ###
E_tot= -499.84154693
Converge= YES
### JOB TYPE = RESP_GSGRAD ###
Energy= -499.841546925072
1 0.0016714972 0.0041574983 -0.0000013445
2 -0.0002556962 -0.0006880567 0.0000402277
3 -0.0002218807 -0.0006861734 -0.0000225761
4 -0.0003229876 -0.0006350885 -0.0000059774
5 -0.0008670369 -0.0021403962 -0.0000084046
可以看到BDFOPT调用了BDF程序本身,来计算初猜结构下分子的SCF能量和梯度。SCF和梯度计算的详细输出在 .out.tmp 文件中,而 .out 文件仅摘取能量值、梯度值,以及SCF是否收敛等信息。其中,能量的单位为Hartree,梯度的单位为Hartree/Bohr。
solver = 1 表示使用BDF自身的优化器,在冗余内坐标下进行结构的优化。
为了产生下一步的分子结构,必须先产生分子的冗余内坐标。
因此在第一步结构优化时,输出文件还会给出各个冗余内坐标的定义(即参与形成相应的键、键角、二面角的原子编号),
以及它们的值(键长的单位为埃,键角、二面角的单位为度):
|******************************************************************************|
Redundant internal coordinates on Angstrom/Degree
Name Definition Value Constraint
R1 1 2 1.0700 No
R2 1 3 1.0700 No
R3 1 4 1.0700 No
R4 1 5 1.7600 No
A1 2 1 3 109.47 No
A2 2 1 4 109.47 No
A3 2 1 5 109.47 No
A4 3 1 4 109.47 No
A5 3 1 5 109.47 No
A6 4 1 5 109.47 No
D1 4 1 3 2 -120.00 No
D2 5 1 3 2 120.00 No
D3 2 1 4 3 -120.00 No
D4 3 1 4 2 120.00 No
D5 5 1 4 2 -120.00 No
D6 5 1 4 3 120.00 No
D7 2 1 5 3 120.00 No
D8 2 1 5 4 -120.00 No
D9 3 1 5 2 -120.00 No
D10 3 1 5 4 120.00 No
D11 4 1 5 2 120.00 No
D12 4 1 5 3 -120.00 No
|******************************************************************************|
待分子结构更新完成后,程序计算梯度以及几何步长的大小,判断结构优化是否收敛:
Force-RMS Force-Max Step-RMS Step-Max
Conv. tolerance : 0.2000E-03 0.3000E-03 0.8000E-03 0.1200E-02
Current values : 0.8833E-02 0.2235E-01 0.2445E-01 0.5934E-01
Geom. converge : No No No No
仅当均方根力(Force-RMS)、最大力(Force-Max)、均方根步长(Step-RMS)、最大步长(Step-Max)的当前值均小于对应的收敛限的时候(也即 Geom. converge 栏均为Yes),程序才认为结构优化收敛。对于本算例,结构优化在第5步时收敛,此时输出信息不仅包含各收敛判据的值,还会明确告知用户几何优化已收敛,并分别以笛卡尔坐标和内坐标的形式打印收敛的分子结构:
Good Job, Geometry Optimization converged in 5 iterations!
Molecular Cartesian Coordinates (X,Y,Z) in Angstrom :
C -0.93557703 0.15971089 0.58828595
H -1.71170348 -0.52644336 0.21665897
H -1.26240747 1.20299703 0.46170050
H -0.72835075 -0.04452039 1.64971607
Cl 0.56770184 -0.09691413 -0.35697029
Force-RMS Force-Max Step-RMS Step-Max
Conv. tolerance : 0.2000E-03 0.3000E-03 0.8000E-03 0.1200E-02
Current values : 0.1736E-05 0.4355E-05 0.3555E-04 0.6607E-04
Geom. converge : Yes Yes Yes Yes
Print Redundant internal coordinates of the converged geometry
|******************************************************************************|
Redundant internal coordinates on Angstrom/Degree
Name Definition Value Constraint
R1 1 2 1.1006 No
R2 1 3 1.1006 No
R3 1 4 1.1006 No
R4 1 5 1.7942 No
A1 2 1 3 110.04 No
A2 2 1 4 110.04 No
A3 2 1 5 108.89 No
A4 3 1 4 110.04 No
A5 3 1 5 108.89 No
A6 4 1 5 108.89 No
D1 4 1 3 2 -121.43 No
D2 5 1 3 2 119.28 No
D3 2 1 4 3 -121.43 No
D4 3 1 4 2 121.43 No
D5 5 1 4 2 -119.28 No
D6 5 1 4 3 119.29 No
D7 2 1 5 3 120.00 No
D8 2 1 5 4 -120.00 No
D9 3 1 5 2 -120.00 No
D10 3 1 5 4 120.00 No
D11 4 1 5 2 120.00 No
D12 4 1 5 3 -120.00 No
|******************************************************************************|
注意此处的均方根力和均方根步长的收敛限可以分别通过 tolgrad 和 tolstep 关键词来设定,程序自动根据设定值来调整最大力和最大步长的收敛限;当使用DL-FIND库时(见后),还可用 tolene 指定能量收敛限。不过一般不建议用户自行调整收敛限。
与此同时,程序还会产生后缀为 .optgeom 的文件,其内容是优化后的分子结构的笛卡尔坐标(若是一般的单点计算,则为当前结构在标准取向下的笛卡尔坐标),但单位为Bohr而非Angstrom:
GEOM
C -0.7303234729 -2.0107211546 -0.0000057534
H -0.5801408002 -2.7816264533 1.9257943885
H 0.4173171420 -3.1440530286 -1.3130342173
H -2.7178161476 -2.0052051760 -0.6126883555
Cl 0.4272106261 1.1761889168 -0.0000021938
.optgeom 文件可以用 $BDFHOME/sbin/ 下的工具 optgeom2xyz.py 转为xyz格式,从而可以在支持xyz格式的任何可视化软件里观看优化后的分子结构。例如待转换的文件名为filename.optgeom,则在命令行执行:(注意必须先设定环境变量$BDFHOME,或手动用BDF文件夹的路径替代下述命令里的$BDFHOME)
$BDFHOME/sbin/optgeom2xyz.py filename
即可在当前目录下得到filename.xyz。
备注
由以上步骤得到的.xyz文件里的坐标,虽然单位为Angstrom,但数值未必和.out文件里的坐标相同,这是正常现象,原因是两个文件的分子朝向以及坐标原点不同。同理,.optgeom文件里的坐标和.out文件里的坐标也并非简单的单位换算关系,而是可能还相差一个平动和一个转动。
最后顺便指出,当分子里有的键角接近或等于180度时,基于冗余内坐标的优化算法经常会出现数值不稳定性问题,导致优化无法继续进行。因此程序在产生冗余内坐标时,会尽量避免选取接近或等于180度的键角。但是即便如此,还是有可能有本来远小于180度的键角在优化过程中接近180度,导致数值不稳定问题,此时程序会自动重新构建冗余内坐标并自动重启优化,并输出如下信息:
Something wrong in getting dihedral!
This is probably because one or more angles have become linear.
--- Restarting optimizer ... (10 attempt(s) remaining) ---
由输出信息可知,程序总共允许优化器重启10次。一般情况下,仅需重启1~2次优化器即可成功优化得到结构,但极少数情况下,10次重启机会用完以后仍然会遇到键角接近180度的问题,此时程序报错退出:
bdfopt_bdf: fatal: the maximum number of restarts has been reached
Please check if your geometry makes sense!
此时用户应检查.optgeom文件里的当前坐标是否合理,如合理,则可改用DL-Find优化器,在直角坐标下进行优化,详情请参见 几何优化不收敛的解决方法 。
频率计算:\(\ce{CH3Cl}\) 在平衡结构下的谐振频率及热化学量的计算
结构优化收敛后,即可进行频率分析。准备以下输入文件:
$compass
title
CH3Cl freq
basis
def2-SV(P)
geometry
C -0.93557703 0.15971089 0.58828595
H -1.71170348 -0.52644336 0.21665897
H -1.26240747 1.20299703 0.46170050
H -0.72835075 -0.04452039 1.64971607
Cl 0.56770184 -0.09691413 -0.35697029
end geometry
$end
$bdfopt
hess
only
$end
$xuanyuan
$end
$scf
rks
dft
b3lyp
$end
$resp
geom
$end
其中分子结构为上述结构优化任务得到的收敛的结构。注意我们在BDFOPT模块中添加了 hess only ,其中 hess 代表计算(数值)Hessian,而 only 的含义将在后续章节详述。对于基态DFT计算,程序进行解析Hessian计算,在.out文件里的输出较少,Hessian的计算细节输出到.out.tmp文件中。对于TDDFT等暂不支持解析Hessian的理论级别,如TDDFT,程序将自动改为数值Hessian计算,此时程序将分子中的每个原子分别向x轴正方向、x轴负方向、y轴正方向、y轴负方向、z轴正方向、z轴负方向进行扰动,并计算扰动结构下的梯度,如:
Displacing atom 1 (+x)...
### [bdf_single_point] ### nstate= 1
Do not allow rotation to standard orientation.
BDFOPT run - details of gradient calculations will be written
into .out.tmp file.
...
### JOB TYPE = SCF ###
E_tot= -499.84157717
Converge= YES
### JOB TYPE = RESP_GSGRAD ###
Energy= -499.841577166026
1 0.0005433780 -0.0000683370 -0.0000066851
2 -0.0000516384 0.0000136326 -0.0000206081
3 -0.0001360377 0.0000872513 0.0000990006
4 -0.0003058645 0.0000115926 -0.0000775624
5 -0.0000498284 -0.0000354732 0.0000023346
备注
因扰动结构会破坏分子的点群对称性,所以即便用户输入的分子存在点群对称性,计算也会自动改为在C(1)群下进行。如果用户希望指定每个不可约表示下的轨道占据数,或希望计算某个特定不可约表示下的某个激发态的数值频率,则用户必须先单独做一个保持点群对称性的单点计算,根据单点计算的结果手动指认用户希望占据的轨道或希望计算的激发态对应于C(1)群下的哪个/哪些轨道或激发态,再根据指认结果撰写C(1)群下的数值频率计算输入文件。
若体系的原子数为N,则共需计算6N个梯度。然而实际上程序还会顺便计算未扰动的结构的梯度,以供用户检查前述结构优化是否确实已经收敛,因此程序实际共计算6N+1个梯度。最后程序通过有限差分方法得到体系的Hessian:
|--------------------------------------------------------------------------------|
Molecular Hessian - Numerical Hessian (BDFOPT)
1 2 3 4 5 6
1 0.5443095266 -0.0744293569 -0.0000240515 -0.0527420800 0.0127361607 -0.0209022664
2 -0.0744293569 0.3693301504 -0.0000259750 0.0124150102 -0.0755387479 0.0935518380
3 -0.0000240515 -0.0000259750 0.5717632089 -0.0213157291 0.0924260912 -0.2929392390
4 -0.0527420800 0.0124150102 -0.0213157291 0.0479752418 -0.0069459473 0.0239610358
5 0.0127361607 -0.0755387479 0.0924260912 -0.0069459473 0.0867377886 -0.0978524147
6 -0.0209022664 0.0935518380 -0.2929392390 0.0239610358 -0.0978524147 0.3068416997
7 -0.1367366097 0.0869338594 0.0987840786 0.0031968314 -0.0034098009 -0.0016497426
8 0.0869913627 -0.1185605401 -0.0945336434 -0.0070787068 0.0099076105 0.0045621064
9 0.0986508197 -0.0953400774 -0.1659434327 0.0163191407 -0.0140134399 -0.0166739137
10 -0.3054590932 0.0111756577 -0.0774713107 0.0016297078 0.0019657599 -0.0021771884
11 0.0112823039 -0.0407134661 0.0021058508 0.0106623780 0.0018506067 0.0005120364
12 -0.0775840113 0.0018141942 -0.0759448618 -0.0275602878 0.0006820252 -0.0059830018
13 -0.0486857506 -0.0362556088 0.0000641125 -0.0000787206 -0.0045253276 0.0011289985
14 -0.0360823429 -0.1334063062 0.0000148321 -0.0091074064 -0.0228930763 -0.0010993076
15 0.0001686252 0.0004961854 -0.0352553706 0.0084860406 0.0189117305 0.0079690194
7 8 9 10 11 12
1 -0.1367366097 0.0869913627 0.0986508197 -0.3054590932 0.0112823039 -0.0775840113
2 0.0869338594 -0.1185605401 -0.0953400774 0.0111756577 -0.0407134661 0.0018141942
3 0.0987840786 -0.0945336434 -0.1659434327 -0.0774713107 0.0021058508 -0.0759448618
4 0.0031968314 -0.0070787068 0.0163191407 0.0016297078 0.0106623780 -0.0275602878
5 -0.0034098009 0.0099076105 -0.0140134399 0.0019657599 0.0018506067 0.0006820252
6 -0.0016497426 0.0045621064 -0.0166739137 -0.0021771884 0.0005120364 -0.0059830018
7 0.1402213115 -0.0861503922 -0.1081442631 -0.0130805143 0.0143574755 0.0192323598
8 -0.0861503922 0.1322736798 0.1009922720 0.0016534140 0.0024111759 0.0011733340
9 -0.1081442631 0.1009922720 0.1688786678 -0.0038440081 0.0072277457 0.0091535975
10 -0.0130805143 0.0016534140 -0.0038440081 0.3186765202 -0.0079165663 0.0838593213
11 0.0143574755 0.0024111759 0.0072277457 -0.0079165663 0.0509206668 -0.0029665370
12 0.0192323598 0.0011733340 0.0091535975 0.0838593213 -0.0029665370 0.0707430980
13 0.0064620333 0.0044161973 -0.0031236007 -0.0026369496 -0.0283860480 0.0017966445
14 -0.0119743475 -0.0258901434 0.0013817613 -0.0066143965 -0.0145372292 -0.0006143935
15 -0.0078330845 -0.0126024853 0.0040383425 -0.0008566397 -0.0068931757 0.0018028482
13 14 15
1 -0.0486857506 -0.0360823429 0.0001686252
2 -0.0362556088 -0.1334063062 0.0004961854
3 0.0000641125 0.0000148321 -0.0352553706
4 -0.0000787206 -0.0091074064 0.0084860406
5 -0.0045253276 -0.0228930763 0.0189117305
6 0.0011289985 -0.0010993076 0.0079690194
7 0.0064620333 -0.0119743475 -0.0078330845
8 0.0044161973 -0.0258901434 -0.0126024853
9 -0.0031236007 0.0013817613 0.0040383425
10 -0.0026369496 -0.0066143965 -0.0008566397
11 -0.0283860480 -0.0145372292 -0.0068931757
12 0.0017966445 -0.0006143935 0.0018028482
13 0.0450796910 0.0642866688 0.0000350066
14 0.0642866688 0.1954779468 0.0000894464
15 0.0000350066 0.0000894464 0.0213253497
|--------------------------------------------------------------------------------|
其中第3N+1(3N+2、3N+3)行对应第N个原子的x(y、z)坐标,第3N+1(3N+2、3N+3)列同理。
接下来BDF调用UniMoVib程序进行频率和热力学量的计算。首先是振动所属不可约表示、振动频率、约化质量、力常数和简正模的结果:
************************************
*** Properties of Normal Modes ***
************************************
Results of vibrations:
Normal frequencies (cm^-1), reduced masses (AMU), force constants (mDyn/A)
1 2 3
Irreps A1 E E
Frequencies 733.9170 1020.5018 1021.2363
Reduced masses 7.2079 1.1701 1.1699
Force constants 2.2875 0.7179 0.7189
Atom ZA X Y Z X Y Z X Y Z
1 6 -0.21108 -0.57499 -0.00106 -0.04882 0.01679 0.10300 0.09664 -0.03546 0.05161
2 1 -0.13918 -0.40351 0.04884 -0.06700 -0.59986 -0.13376 -0.37214 -0.36766 -0.03443
3 1 -0.11370 -0.42014 -0.03047 0.26496 0.65294 -0.15254 -0.28591 -0.18743 -0.15504
4 1 -0.19549 -0.38777 -0.01079 0.05490 -0.14087 -0.24770 0.15594 0.73490 -0.07808
5 17 0.08533 0.23216 0.00014 0.00947 -0.00323 -0.01995 -0.01869 0.00699 -0.01000
其中各振动模式是按振动频率从小到大的顺序排列的,而虚频排在所有实频的前面,因此只需检查前几个频率,即可得知虚频的数目。接下来打印热化学分析结果:
*********************************************
*** Thermal Contributions to Energies ***
*********************************************
Molecular mass : 49.987388 AMU
Electronic total energy : -499.841576 Hartree
Scaling factor of Freq. : 1.000000
Tolerance of scaling : 0.000000 cm^-1
Rotational symmetry number: 3
The C3v point group is used to calculate rotational entropy.
Principal axes and moments of inertia in atomic units:
1 2 3
Eigenvalues -- 11.700793 137.571621 137.571665
X 0.345094 0.938568 -0.000000
Y 0.938568 -0.345094 -0.000000
Z 0.000000 0.000000 1.000000
Rotational temperatures 7.402388 0.629591 0.629591 Kelvin
Rot. constants A, B, C 5.144924 0.437588 0.437588 cm^-1
154.240933 13.118557 13.118553 GHz
# 1 Temperature = 298.15000 Kelvin Pressure = 1.00000 Atm
====================================================================================
Thermal correction energies Hartree kcal/mol
Zero-point Energy : 0.037519 23.543449
Thermal correction to Energy : 0.040539 25.438450
Thermal correction to Enthalpy : 0.041483 26.030936
Thermal correction to Gibbs Free Energy : 0.014881 9.338203
Sum of electronic and zero-point Energies : -499.804057
Sum of electronic and thermal Energies : -499.801038
Sum of electronic and thermal Enthalpies : -499.800093
Sum of electronic and thermal Free Energies: -499.826695
====================================================================================
用户可根据需要读取零点能、焓、Gibbs自由能等数据。注意以上所有热力学量是在以下各个假设下得到的:
频率校正因子为 1.0;
温度为 298.15 K;
压强为 1 atm;
电子态的简并度为1。
如用户的计算不属于以上情形,可以通过一系列关键词进行指定,如以下的写法代表频率校正因子为0.98,温度为373.15 K,压强为2 atm,电子态的简并度为2:
$bdfopt
hess
only
scale
0.98
temp
373.15
press
2.0
ndeg
2
$end
其中尤其需要注意的是电子态的简并度,对于非相对论或标量相对论计算,且电子态不存在空间简并性的情形,电子态的简并度等于自旋多重度(2S+1);对于存在空间简并性的电子态,还应乘上电子态的空间简并度,也即电子波函数的空间部分所属不可约表示的维数。至于考虑了旋轨耦合的相对论性计算(如TDDFT-SOC计算),则应将自旋多重度替换为相应旋量态的简并度(2J+1)。
在热化学数据之后,程序会检查梯度,根据内置的比较宽松的阈值,确认是否为驻点结构。与结构优化步骤不同,这里检测的是直角坐标梯度而不是内坐标梯度, 因为后者在个别情况下存在数值问题,容易导致误判。
有时因SCF不收敛或其他外在原因,导致频率计算中断,此时可在BDFOPT模块里加入 restarthess 关键词进行断点续算,节省计算时间,如:
$bdfopt
hess
only
restarthess
$end
此外值得注意的是,可以在同一个BDF任务里依次实现结构优化与频率分析(即所谓的opt+freq计算),而无需单独编写两个输入文件。为此只需将BDFOPT模块的输入改为:
$bdfopt
solver
1
hess
final
$end
其中final表示在结构优化成功结束后才进行数值Hessian计算;若结构优化不收敛,则程序直接报错退出,而不进行Hessian及频率、热力学量的计算。由此可以看出,前述的频率计算输入文件中的only,即为只进行频率计算而不进行结构优化之意。
备注
虽然opt+freq计算中的结构优化步骤支持在非C(1)点群下计算,但数值频率计算步骤仍然必须在C(1)群下计算。所以如果用户计算的分子具有点群对称性,且希望指定各个不可约表示的轨道占据数或指定优化某个特定不可约表示下的激发态,则必须先做结构优化,再根据前述步骤手动指认相应的轨道/激发态对应于C(1)群下的哪些轨道/激发态,再在C(1)群下进行数值频率计算,而不能直接做opt+freq计算。
对于大分子(例如100个原子以上)、柔性分子,以及非共价相互作用比较重要的体系的自由能、熵计算,建议打开QRRHO,有助于提高精度,方法为在
$bdfopt块中增加QRRHO关键字。对于刚性小体系,QRRHO对计算结果几乎没有影响。开启QRRHO后,自由能、熵结果与ORCA、Turbomole可比,但与Gaussian不可比。若所研究的课题涉及至少一个非共价相互作用很重要的体系的自由能计算,且不需要计算结果与Gaussian可比,则建议总是打开QRRHO。注意若同一个工作既涉及需要开QRRHO的计算,又涉及不是一定要开QRRHO的计算,则应当统一开QRRHO,否则属于理论级别不统一,结果不可比。解析Hessian计算的内存并非完全由环境变量
OMP_STACKSIZE控制(后者只控制每个核的栈内存,而不控制堆内存),而是还由resp模块的maxmem关键词控制(该关键词控制的是所有核的总堆内存,在2025年7月及之后的版本中支持)。堆内存与栈内存之和必须小于节点总物理内存,考虑到内存估计的误差及同一个节点上的其他进程消耗的内存,建议小于节点总物理内存的80%。也即:OMP_STACKSIZE*OMP_NUM_THREADS + maxmem <= 物理内存*80%。对于解析Hessian计算,建议OMP_STACKSIZE*OMP_NUM_THREADS为maxmem的1/3~1/10左右,但若这使得OMP_STACKSIZE小于1G,则设置OMP_STACKSIZE=1G。默认情况下,程序会用用户分配的所有的核计算一个梯度,然后再用所有的核计算下一个梯度,等等。当计算所用核数较多(如大于16),且体系较大(如多于50个原子)时,用完美并行(embarrassingly parallel)的方法进行梯度计算往往是更高效的。此时可以在bdfopt模块中指定
ParHess关键字,此时默认会开启OMP_NUM_THREADS个进程,每个进程计算一个梯度,且每个梯度计算仅用一个核,仅当计算接近结束、剩余待计算梯度数目小于OMP_NUM_THREADS时,每个梯度计算用的核数才会动态增加。当设定NCorePerGrad关键词时,每个梯度计算使用NCorePerGrad个核,有助于改善计算末期的负载均衡,适用于计算所用核数非常多而体系不是很大的情形。注意:ParHess关键字仅在2025年7月及以后的BDF版本中可用,且要求用户安装Python3、NumPy和SciPy。
O1NumHess:仅用O(1)个梯度进行近似的数值频率计算
传统的数值Hessian算法需要计算6N个梯度,其中N为原子数,当体系较大时计算量非常可观。之所以需要O(N)个梯度,是因为Hessian包含O(N^2)个矩阵元,而每个梯度仅包含O(N)个数字,因此从信息论角度讲,需要O(N)个梯度才能唯一确定Hessian的所有矩阵元。然而,大体系的Hessian通常是稀疏的,即使不是稀疏的(例如对于HOMO-LUMO gap较小的体系或激发态,Hessian的稀疏性往往较差),其非对角块也往往有一定结构,即呈现低秩性。因此,仅需O(N)个参数即可以较高精度近似描述整个Hessian,也就是说只需要O(1)个梯度,原则上即可近似地计算出整个Hessian,只不过需要较复杂的数据处理方法。
自2025年7月起,BDF接入了O1NumHess库 [79] ,支持用O(1)个梯度计算数值Hessian。该部分代码除作为BDF的一部分发行外,也已单独在GitHub上开源(https://github.com/ilcpm/O1NumHess、https://github.com/ilcpm/O1NumHess_QC/),可结合更早版本的BDF使用,使用方法参见http://bbs.keinsci.com/thread-55109-1-3.html。不管对于多大的体系,O1NumHess的计算量都比传统数值Hessian算法小50%以上;对于足够大的体系,O1NumHess仅需计算100个左右的梯度,对于150原子以上的体系其计算量可小至传统数值Hessian的1/10左右。因此O1NumHess的主要应用场合是在程序暂不支持解析Hessian的情形下,例如计算TDDFT Hessian,或在开启MPEC+COSX的情况下计算基态Hessian的情况下,此时打开O1NumHess可以大大节约时间。当所用理论方法支持解析Hessian时,虽然对于小体系,或在内存充足的条件下,O1NumHess的计算速度往往较解析Hessian更慢,但是在内存受限或并行核数较多的情况下,O1NumHess的计算速度是有可能较解析Hessian更快的。精度方面,默认设置下,O1NumHess的平均振动频率误差一般为2~5 cm-1,反应Gibbs自由能误差一般在1 kcal/mol左右。
使用O1NumHess算法的方法为在普通Hessian输入文件的基础上,在bdfopt模块中添加 o1numhess 关键词。例如以下输入文件在HF/STO-3G级别下计算1-辛醇的Hessian:
$compass
Title
1-Oct-OH
Basis
STO-3G
Geometry
C 0.35104892 5.02179154 0.00000000
H 0.96877389 5.02880594 -0.87365134
H 0.96877389 5.02880594 0.87365134
H -0.27659591 5.88837215 0.00000000
C -0.52373598 3.75437236 0.00000000
H -1.14146094 3.74735795 0.87365134
H -1.14146094 3.74735795 -0.87365134
C 0.37960332 2.50714418 0.00000000
H 0.99732828 2.51415859 -0.87365134
H 0.99732828 2.51415859 0.87365134
C -0.49518158 1.23972500 0.00000000
H -1.11290655 1.23271060 -0.87365134
H -1.11290655 1.23271060 0.87365134
C 0.40815771 -0.00750317 0.00000000
H 1.02588267 -0.00048876 0.87365134
H 1.02588267 -0.00048876 -0.87365134
C -0.46662719 -1.27492235 0.00000000
H -1.08435216 -1.28193676 -0.87365134
H -1.08435216 -1.28193676 0.87365134
C 0.43671210 -2.52215052 0.00000000
H 1.05443707 -2.51513612 0.87365134
H 1.05443707 -2.51513612 -0.87365134
C -0.43807280 -3.78956970 0.00000000
H -1.05579776 -3.79658411 -0.87365134
H -1.05579776 -3.79658411 0.87365134
O 0.40074226 -4.94771015 0.00000000
H -0.14457820 -5.73778964 0.00000000
End Geometry
nosym
norotate
$end
$bdfopt
hess
only
qrrho # not necessary for o1numhess, but necessary for large, esp. noncovalent systems
o1numhess
ncorepergrad
2 # number of cores per gradient calculation, default: 1
$end
$xuanyuan
$end
$scf
RHF
$end
$resp
geom
$end
注意这里还涉及一个新的关键词 ncorepergrad ,该关键词指定每个梯度计算用多少个核。例如当 OMP_NUM_THREADS 为8,而 ncorepergrad 为2时,表示:程序同时进行4个梯度计算,每个计算使用2个核;但当剩余待计算的梯度数目不到4个时,每个梯度计算可能会使用2个以上的核数,但所有梯度计算的总核数仍然不超过8个。 ncorepergrad 的设置只会影响计算速度,而不会影响结果的正确性。 ncorepergrad 的最优取值应使得OMP_NUM_THREADS除以 ncorepergrad 约为预期需要计算的梯度数的1/5~1/10左右(预期需要计算的梯度数按min(100, 3N-4)估算,N为体系原子数),但若这使得 ncorepergrad 小于1,则取 ncorepergrad=1 。该设置一般可实现负载均衡和并行效率的较好平衡。
计算分为三个阶段,第一阶段进行大部分梯度的计算:
Stage 1: Initial estimation of the Hessian
Start O1NumHess run...
7 displacement directions given on input
44 displacement directions generated
Total number of displacement directions: 51
46 gradients will be calculated
Start gradient calculations...
Gradient calculations finished
Enter _genODLRHessian
Enter _genLocalHessian
Successful termination of _genLocalHessian, time = 0.08 sec
Error norm of the predicted gradient: 6.71e-03
The gradients cannot be exactly reproduced by a symmetric Hessian.
Exit _genODLRHessian
Successful termination of _genODLRHessian, time = 0.08 sec
Error norm of the predicted gradient: 6.06e-03
Stage 1 done, total time: 93.50 sec
可以看到程序产生了51个扰动的方向,但有的扰动方向下需要进行双向数值差分,大部分扰动方向下只需进行单向数值差分,且有的扰动方向对Hessian的贡献无需计算即可得知(即平动、转动)。因此该步骤实际仅需计算46个梯度。得到梯度后,程序调用 _genODLRHessian ,在假设Hessian的非对角块低秩的情况下,对Hessian矩阵元进行估算。接下来是第二阶段,程序将Hessian对角化:
Stage 2: Check imaginary modes
- 12 imaginary mode(s) found
Stage 2 done, total time: 0.00 sec
因计算误差,Hessian包含大量的小虚频。接下来在第三阶段中,程序在这些虚频的方向上进行更多的梯度计算,以确认这些虚频是否真的存在:
Stage 3: Displace along the imaginary modes
Start O1NumHess run...
63 displacement directions given on input
0 displacement directions generated
Total number of displacement directions: 63
12 gradients will be calculated
Start gradient calculations...
Gradient calculations finished
Enter _genODLRHessian
Enter _genLocalHessian
Successful termination of _genLocalHessian, time = 0.03 sec
Error norm of the predicted gradient: 1.55e-02
Successful termination of _genODLRHessian, time = 0.05 sec
Error norm of the predicted gradient: 1.45e-02
Stage 3 done, total time: 23.31 sec
Exit runO1NumHess
BDF numerical Hessian done, total time: 117.07 sec
因此,整个计算调用的梯度计算数目为46+12=58个。因体系含有27个原子,传统数值Hessian算法将需要27*6=162个梯度计算,也即O1NumHess相比传统数值Hessian算法的加速比为2.8。对于更大的体系,加速比一般更加显著。
与其他Hessian计算相同,输出文件接下来打印Hessian矩阵,频率分析结果,及热化学分析结果。其中热化学分析结果为
====================================================================================
Thermal correction energies Hartree kcal/mol
Zero-point Energy : 0.299658 188.037987
Thermal correction to Energy : 0.305404 191.644150
Thermal correction to Enthalpy : 0.306349 192.236635
Thermal correction to Gibbs Free Energy : 0.268255 168.332795
Sum of electronic and zero-point Energies : -383.302117
Sum of electronic and thermal Energies : -383.296370
Sum of electronic and thermal Enthalpies : -383.295426
Sum of electronic and thermal Free Energies: -383.333519
====================================================================================
对比解析Hessian的结果:
====================================================================================
Thermal correction energies Hartree kcal/mol
Zero-point Energy : 0.299816 188.137300
Thermal correction to Energy : 0.305553 191.737267
Thermal correction to Enthalpy : 0.306497 192.329753
Thermal correction to Gibbs Free Energy : 0.268419 168.435170
Sum of electronic and zero-point Energies : -383.301959
Sum of electronic and thermal Energies : -383.296222
Sum of electronic and thermal Enthalpies : -383.295278
Sum of electronic and thermal Free Energies: -383.333356
====================================================================================
可以看到零点能、自由能误差仅0.1 kcal/mol。因一般关心零点能、自由能的差而非绝对数值,在作差时还可以实现误差抵消,导致最终结果的误差更低。
备注
O1NumHess需要用户安装Python3(3.6或以上版本)、NumPy和SciPy。若用户在登录节点上安装了Python3、NumPy和SciPy,但没有在计算节点上安装,需要确认计算节点是否能正确调用这三者。
计算会产生大量的形如$BDFTASK_000.*, $BDFTASK_001.*等的文件(其中数字部分未必是3位),默认情况下程序会自动删除这些文件,其中较大的文件(如波函数文件等)在计算完对应的梯度后立即删除,较小的文件在计算完全部梯度后才删除。例外是当某个梯度计算出现SCF不收敛等问题,导致报错时,对应的输出文件、波函数文件等不会删除,方便用户排查不收敛的原因。若要求即使在计算正常结束的情况下也保留这些文件,可在 $bdfopt 输入块中加入 nodeletegradfiles 关键词。
由以上算例和TDDFT梯度计算的输入文件写法,不难推知用O1NumHess计算TDDFT Hessian的输入文件写法,此处不另给算例。其中TDDFT Hessian除用于计算热化学校正量、激发态虚频数目外,还可结合MOMAP用于计算振动分辨光谱、跃迁速率等。
O1NumHess也可用于计算结构优化的初始Hessian,或在结构优化结束后计算Hessian(即"opt freq"计算)。
过渡态结构优化:HCN/HNC异构反应的过渡态优化和频率计算
准备以下输入文件:
$compass
title
HCN <-> HNC transition state
basis
def2-SVP
geometry
C 0.00000000 0.00000000 0.00000000
N 0.00000000 0.00000000 1.14838000
H 1.58536000 0.00000000 1.14838000
end geometry
$end
$bdfopt
solver
1
hess
init+final
iopt
10
$end
$xuanyuan
$end
$scf
rks
dft
b3lyp
$end
$resp
geom
$end
其中 iopt 10 表示优化过渡态。
无论是优化极小值点结构,还是优化过渡态,程序都必须在第一步结构优化之前产生一个初始的Hessian,以备后续结构优化步骤使用。一般而言,初始Hessian应当与初始结构下的精确Hessian定性符合,尤其是虚频数目必须一致。对于极小值点的优化,这个要求很容易满足,即便是分子力学级别的Hessian(所谓“模型Hessian”)也能做到和精确Hessian定性一致,因此此时程序以模型Hessian为初始Hessian,而无需计算精确Hessian。然而对于过渡态优化,模型Hessian一般不存在虚频,因此必须产生精确Hessian作为初始Hessian。以上输入文件的 hess init+final 即表示既产生初始Hessian以备过渡态优化需要(此Hessian因为不是在梯度为0的结构上计算的,频率及热化学量没有明确物理意义,因此仅计算Hessian而不做频率分析),又在结构优化收敛后再次进行Hessian计算,以得到频率分析结果。也可将 init+final 替换为 init ,即只产生初始Hessian,而结构优化收敛后不再次计算Hessian,但因过渡态优化(乃至所有结构优化任务)一般需要检验最终收敛的结构的虚频数目,因此不建议省略final关键词。
计算的输出与优化极小值点结构类似。最后频率分析时可以看到收敛的结构有且仅有一个虚频(-1104 \(\rm cm^{-1}\)):
Results of vibrations:
Normal frequencies (cm^-1), reduced masses (AMU), force constants (mDyn/A)
1 2 3
Irreps A' A' A'
Frequencies -1104.1414 2092.7239 2631.2601
Reduced masses 1.1680 11.9757 1.0591
Force constants -0.8389 30.9012 4.3205
Atom ZA X Y Z X Y Z X Y Z
1 6 0.04309 0.07860 0.00000 0.71560 0.09001 0.00000 -0.00274 -0.06631 0.00000
2 7 0.03452 -0.06617 0.00000 -0.62958 -0.08802 0.00000 0.00688 -0.01481 0.00000
3 1 -0.99304 -0.01621 0.00000 0.22954 0.15167 0.00000 -0.06313 0.99566 0.00000
代表确实找到了过渡态。
在以上计算中,初始Hessian的理论级别与过渡态优化的理论级别一致。因初始Hessian只需定性正确即可,实际计算中可以在另一个较低的级别下计算初始Hessian,再在较高理论级别下优化过渡态。仍以以上算例为例,假如我们想在HF/STO-3G级别下计算初始Hessian,而在B3LYP/def2-SVP级别下优化过渡态,可以按照以下步骤进行:
(1)准备以下输入文件,命名为 HCN-inithess.inp :
$compass
title
HCN <-> HNC transition state, initial Hessian
basis
STO-3G
geometry
C 0.00000000 0.00000000 0.00000000
N 0.00000000 0.00000000 1.14838000
H 1.58536000 0.00000000 1.14838000
end geometry
$end
$bdfopt
hess
only
$end
$xuanyuan
$end
$scf
rhf
$end
$resp
geom
$end
(2)用BDF运行该输入文件,得到Hessian文件 HCN-inithess.hess ;
(3)将 HCN-inithess.hess 复制或重命名为 HCN-optTS.hess ;
(4)准备以下输入文件,命名为 HCN-optTS.inp:
$compass
title
HCN <-> HNC transition state
basis
def2-SVP
geometry
C 0.00000000 0.00000000 0.00000000
N 0.00000000 0.00000000 1.14838000
H 1.58536000 0.00000000 1.14838000
end geometry
$end
$bdfopt
solver
1
hess
init+final
iopt
10
readhess
$end
$xuanyuan
$end
$scf
rks
dft
b3lyp
$end
$resp
geom
$end
其中关键词 readhess 表示读取与该输入文件同名的hess文件(即HCN-optTS.hess)作为初始Hessian。注意尽管该输入文件不会重新计算初始Hessian,仍然需要写 hess init+final 而不是 hess final 。
(5)运行该输入文件即可。
用Dimer方法优化过渡态结构
为了获得过渡态的虚频振动模式,需要执行一次甚至多次的Hessian矩阵计算,这是优化过渡态的标准流程中最耗时的步骤。不过,也有一些过渡态优化方法只需要梯度,不需要计算Hessian矩阵,这就大大提高了计算效率以及量子化学方法的应用范围。 以下介绍的是Dimer方法 [80, 81, 82] 和CI-NEB方法 [83] 。
Dimer方法需要定义两个结构,称为像点(Image),两个像点的间距为一个固定的小值Delta,像点连线称为轴。 在Dimer计算过程中,对两个像点垂直于轴向的力进行最小化(称为旋转Dimer步骤),而在轴向的力进行最大化(称为平移Dimer步骤),最终收敛到过渡态结构。此时,轴向对应着虚频模式,而耗时的Hessian计算被巧妙地避开了。
注意
Dimer方法要调用DL-FIND外部库 [84] (
Solver=0),仅支持L-BFGS优化算法(IOpt=3)。由于DL-FIND与BDF默认的坐标转动有冲突,必须在
compass模块中加上关键词norotate禁止分子转动,或用nosymm关闭对称性;对于双原子和三原子分子,只能用nosymm。此冲突今后会解决。如果在过渡态优化后做频率计算,加上
hess=final。由于Dimer方法不需要Hessian,不要用init+final。
仍然取上一节的例子,加上关键词 dimer 和 nosymm (后者关闭对称性并禁止分子转动),优化方法 iopt 要从10改为默认的3(也可以不指定 iopt ),因为我们不需要计算Hessian。输入文件如下:
$compass
title
HCN <-> HNC transition state
basis
def2-SVP
geometry
C 0.00000000 0.00000000 0.00000000
N 0.00000000 0.00000000 1.14838000
H 1.58536000 0.00000000 1.14838000
end geometry
nosymm
$end
$bdfopt
solver
0
iopt
3
dimer
#hess
# final
$end
$xuanyuan
$end
$scf
rks
dft
b3lyp
$end
$resp
geom
$end
经过14步优化结束:
Testing convergence of dimer midpoint in cycle 14
Energy 0.0000E+00 Target: 1.0000E-06 converged? yes
Max step 1.9375E-04 Target: 8.0000E-04 converged? yes component 4
RMS step 9.0577E-05 Target: 5.3333E-04 converged? yes
Max grad 6.9986E-06 Target: 2.0000E-04 converged? yes component 6
RMS grad 4.0401E-06 Target: 1.3333E-04 converged? yes
Converged!
得到的过渡态总能量为-93.22419648 Hartree,与上一节得到的能量-93.22419582 Hartree非常接近。
Summary printing of molecular geometry and gradient for this step
Atom Coord
C 0.381665 0.002621 0.138107
N -0.079657 -0.020912 1.233092
H 1.283352 0.018291 0.925561
State= 1
Energy= -93.22419612
Gradient=
C 0.00000523 0.00000093 -0.00000335
N 0.00000131 -0.00000022 0.00000700
H -0.00000655 -0.00000070 -0.00000365
如果修改Dimer方法的默认参数,可以把关键词 dimer 改为 Dimer-Block ... End Dimer 输入块。其中的关键词参见BDFOPT模块的说明。
内禀反应坐标(IRC)计算
IRC(Intrinsic Reaction Coordinate)是量子化学研究化学反应的重要概念,它是质权坐标下连接势能面相邻两个极小点的能量最低路径,在不考虑热运动因素下,描述其化学过程中最理想的结构变化轨迹,对讨论微观化学过程至关重要,并且也是验证过渡态正确性的最决定性方法。
BDF的IRC计算是由IRC模块来实现的,使用的算法是支持笛卡尔坐标的Morokuma算法(J. Chem. Phys. 1977, 66, 2153), 该IRC计算需要过渡态结构的力常数,您需要提供优化收敛的过渡态结构 ts.inp 和该结构下的力常数信息 ts.hess ,为了加速IRC计算过程,您还需要提供该结构下收敛的分子轨道信息 ts.scforb 文件,这也意味着需要在BDF的输入文件中添加 guess readmo 这两行。以下对 BDFIRC 模块的输入文件格式举例进行介绍,并对输出文件进行解读。
计算IRC时,需要把 $IRC 模块的参数写在输入文件 3c2o5h.inp 的最前面,并且保留优化过渡态时使用的计算参数,即 $scf 和 $resp 模块需要严格保留。需要说明的是,你需要把ts对应的hess、scforb文件都改名为 3c2o5h.hess, 3c2o5h.scforb,以及把ts收敛的坐标写到 3c2o5h.inp 中。
IRC任务的高级输入为:
$irc
ircpts #反应路径的最大步数
50
ircdir #选择反应路径的方向
0
ircalpha #反应路径的步长参数
0.05
$end
# 下面的参数与计算TS结构时保持一致
$compass
Title
Irc4bdf
Geometry
C -0.81981975 1.01964884 0.22750818
C 0.62889615 1.02275098 -0.13495924
O 1.14639042 -0.35161068 0.02899058
C 0.09835734 -1.20263633 0.05869588
O -1.03508512 -0.71305587 -0.26218679
H -1.52425628 1.52979576 -0.43070061
H -1.07734340 1.04483316 1.29117725
H 0.79223623 1.32207879 -1.18212528
H 1.25263913 1.63815017 0.52777758
H 0.22167944 -2.05610649 0.75197182
End geometry
Basis
CC-PVDZ
$end
$xuanyuan
$end
$scf
uks
guess
readmo
dft
GB3LYP
charge
0
spinmulti
2
mpec+cosx
molden
$end
$resp
Geom
$end
计算结果分析
运行完IRC任务后,BDF会额外生成 3c2o5h.irc 文件和 3c2o5h.trj 文件。
3c2o5h.irc文件保留了每一步计算的信息;3c2o5h.trj文件保留了每一步的轨迹文件。
其中 3c2o5h.irc 会首先给出一些基本的用户设置信息,
IRC wrapper for BDF - version 2023A
Using BDF from: $BDFHOME/sbin/bdfdrv.py
Parameters for this run:
Algorithm: 1
N. Points: 50
Grad. Tol.: 0.0001
Hessian: c3o2h5.hess
Mode: 0
Direction: 1
Alpha: 0.0500
Delta: 0.0500
Damp Factor: 0.0500
Damp Update: 0
Max. Displ.: 0.0100
Guess: c3o2h5.TS.scforb
接下来会有计算的每一帧结构的能量以及RMS Grad,
------------------------------------------------------------------------
Pt. Energy RMS Grad. Damp
-------------------------------------------------------------------------
@ 1 -267.675550488 0.01427 5.00e-02
@ 2 -267.679192880 0.00232 5.00e-02
@ 3 -267.681201395 0.01348 5.00e-02
@ 4 -267.690625817 0.01357 5.00e-02
@ 5 -267.696989174 0.01006 5.00e-02
@ 6 -267.699945911 0.00574 5.00e-02
@ 7 -267.701148839 0.00394 5.00e-02
@ 8 -267.701644838 0.00262 5.00e-02
@ 9 -267.701959150 0.00243 5.00e-02
@ 10 -267.702145221 0.00170 5.00e-02
@ 11 -267.702350215 0.00189 5.00e-02
@ 12 -267.702476410 0.00117 5.00e-02
@ 13 -267.702602274 0.00145 5.00e-02
@ 14 -267.702713773 0.00096 5.00e-02
@ 15 -267.702843816 0.00133 5.00e-02
@ 16 -267.702921941 0.00113 5.00e-02
@ 17 -267.702997038 0.00077 5.00e-02
最后正常收敛的话会出现
----------------------------------------------
-- ENERGY INCREASED --
-- GEOMETRY MIGHT BE --
-- VERY CLOSE TO A MINIMUM --
-- --
-- IRC CALCULATION TERMINATED! --
----------------------------------------------
另外 3c2o5h.trj 就是把每一步的轨迹输出,形式如下,
10
IRC for BDF-irc-f point 1 E= -267.6755505
C -0.774450 1.005021 0.227507
C 0.623378 1.023651 -0.134433
O 1.142071 -0.346726 0.029851
C 0.099686 -1.204880 0.058039
O -1.040129 -0.707532 -0.261197
H -1.538885 1.536311 -0.439412
H -1.084614 1.045208 1.295915
H 0.789098 1.321574 -1.178911
H 1.246292 1.636770 0.526219
H 0.221239 -2.055548 0.752570
10
IRC for BDF-irc-f point 2 E= -267.6791929
C -0.806848 1.013333 0.225962
C 0.635583 1.022502 -0.136684
O 1.144366 -0.348838 0.029978
C 0.099054 -1.205298 0.056237
O -1.037689 -0.705204 -0.259469
H -1.515348 1.520972 -0.422025
H -1.076532 1.043033 1.285840
H 0.789437 1.322192 -1.180942
H 1.248393 1.638410 0.527357
H 0.220724 -2.055404 0.753891
提示
使用IRC模块需要用户安装好numpy;
用户可以把得到的坐标文件输入到Device Studio中,查看轨迹的变化情况,DS平台待更新此功能。
用CI-NEB方法计算最低能量路径和优化过渡态
与原始的拉橡皮筋(Nudged Elastic Band;NEB)方法不同,CI-NEB方法在能量最高点增加了像点爬升(Climbing Image;CI)处理步骤,因此不仅能得到更准确的最低能量(反应)路径,同时还能得到过渡态结构 [83] 。
仍然取上一节HCN异构反应的例子,注意事项参见前面的Dimer方法。
CI-NEB计算需要提供两个端点的坐标,其中第一个端点(这里取反应物HCN)的初始结构在 Compass 模块提供,由于直线形结构不易处理,加了一点扰动。
第二个端点是弯曲结构(经过CI-NEB优化后成为HNC异构体),在 Geometry2 ... End Geometry2 输入块提供。
两套坐标的原子顺序必须一致。输入文件如下:
$compass
basis-block
def2-SVP
end basis
geometry
C 0.0200000 0.0000000 0.0000000
N 0.0000000 -1.1400000 0.0000000
H 0.0000000 1.0500000 0.0000000
end geometry
nosymm
$end
$bdfopt
solver
0
iopt
3
neb-block
crude
nebmode
0
nimage
3
end neb
geometry2
C 0.0000000 0.0000000 0.0000000
N -1.1500000 0.2300000 0.0000000
H -1.6100000 1.1100000 0.0000000
end geometry2
$end
$xuanyuan
$end
$scf
rks
dft
b3lyp
$end
$resp
geom
$end
由于CI-NEB方法的中间像点数越多计算越慢,且增加结构不收敛的几率,因此不建议用太多的中间像点,建议在3至7之间。
如果只关心过渡态,也可以尝试用更少的中间像点,例如1,但对本例不可行,因为反应物和产物是线型结构,而中间像点是弯曲结构,
导致二者的梯度矢量重叠太小,影响结构收敛。
本例用了3个中间像点,由 Nimage 定义,并用 Crude 降低收敛精度,同时对反应物和产物做能量最小化( Nebmode = 0;默认固定不优化)。
除了 Nimage 指定的3个中间像点,还有必须要算的3个像点,因此总像点数为6。其中,1、5像点对应两个端点(即反应物和产物),2、3、4为反应路径上的中间像点。
这5个像点优化到一定程度时,会创建用于爬升步骤的像点6,它最终会优化到过渡态。
经过31步结构优化后,CI-NEB方法找到了最低能量路径:
Testing convergence of NEB climbing image in cycle 31
Energy 7.1900E-07 Target: 4.0000E-05 converged? yes
Max step 1.1193E-03 Target: 5.3333E-03 converged? yes component 8
RMS step 6.5514E-04 Target: 3.5556E-03 converged? yes
Max grad 7.4900E-05 Target: 1.3333E-03 converged? yes component 5
RMS grad 3.6435E-05 Target: 8.8889E-04 converged? yes
Convergence reached
输出文件往前翻,可以看到各个像点(包括反应物、产物、过渡态)的能量总结报告:
NEB Report
Energy F_tang F_perp Dist Angle 1-3 Ang 1-2 Sum
Img 1 -93.3003651 0.00000 0.00000 1.17248 0.00 0.00 63.25 frozen
Img 2 -93.2804319 0.00160 0.00059 1.01235 63.25 86.25 94.29 frozen
Img 3 -93.2270244 -0.00167 0.00049 1.17963 31.04 77.08 80.27 frozen
Img 4 -93.2512597 -0.00248 0.00075 1.42718 49.23 0.00 49.23 frozen
Img 5 -93.2785849 0.00000 0.00000 0.00000 0.00 0.00 0.00 frozen
Cimg 3 -93.2241949 0.00010 0.00007 0.21264 0.00 0.00 0.00
在上面的表格中, Energy 列给出了反应路径上每个像点的能量,其中最后一个 Cimg 表示爬升像点,也就是过渡态, 并且标记了离它最近的像点(本例是3号像点)。 F_tang 列和 F_perp 列给出了每个像点在平行路径和垂直路径方向的受力,原则上应当接近零。 Dist 列、 Angle 列、、 1-3 Ang 列、 1-2 Sum 列给出了路径的特性,分别当前像点为到下一个像点的距离(可以看到NEB的像点经过优化后,不再是等间隔的), 当前像点与上一个像点的角度,下一个像点与上一个像点的角度,以及当前像点和下一个像点的角度之和。 把 Dist 列的数据累加作为横轴,能量作为纵轴,可以绘制NEB反应坐标能量图(这些数据可以在单独的文件nebinfo中找到)。
Cimg 这一行给出的过渡态总能量是-93.2241949 Hartree(DL-FIND最终打印的 Final converged energy 也是这个能量),与之前Dimer方法优化得到的过渡态总能量-93.22419648 Hartree非常接近。
过渡态的直角坐标可以在能量数据之前找到(原子单位),同时也打印在CI-NEB保存的文本文件 neb_0006.xyz (因为过渡态在本例中是第六个像点)的末尾,单位是埃。 其余5个像点的直角坐标保存在文本文件nebpath.xyz中(单位:埃)。 如果必要,也可以用前面介绍的方法对过渡态结构做更严格的优化,这要比在CI-NEB计算中直接用严格收敛标准更加高效。
把每一步优化的像点能量提取出来,画NEB轨迹图如下。作为演示,横轴坐标取自最后一步,但实际上每一步的横轴坐标都会有一些变化。
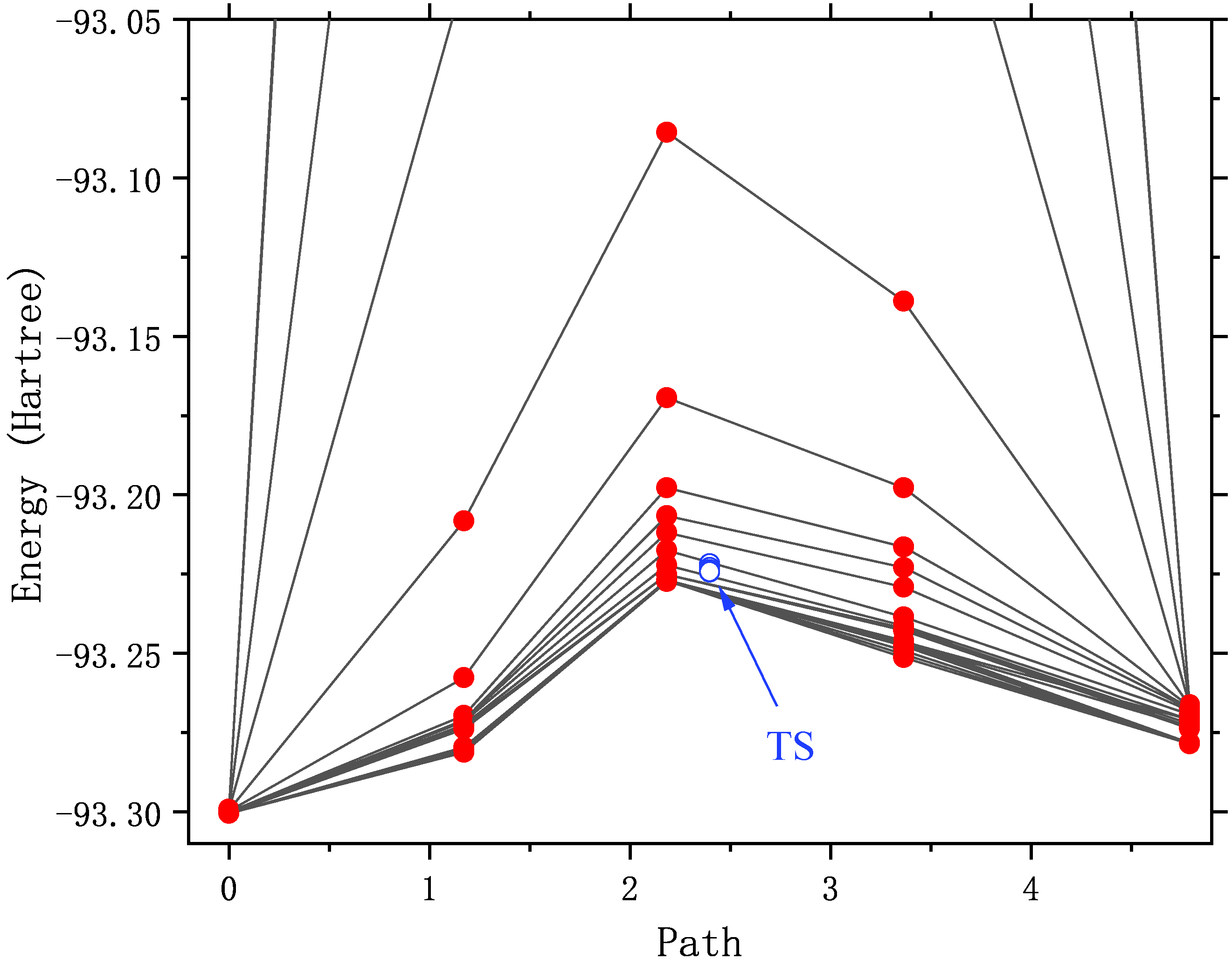
可见随着优化,路径的能量逐渐降低,直至收敛。或许有人注意到了,2至4号像点在最初几轮结构优化中的能量非常高(超出显示范围), 说明这些点的初始结构不太合理。例如,在3号像点的初始结构中,C-N键长仅有0.5埃!不合理的结构不仅会阻碍结构收敛,还破坏SCF收敛, 或者收敛到我们不想要的激发态上。
CI-NEB计算经常不收敛。有以下处理方法:
提取能量最高像点的结构,或者爬升像点的结构,改用之前介绍的过渡态优化方法进行计算。
提取能量最高的两个像点的结构(例如本算例的3、4)作为初始结构,重新做CI-NEB优化,但是此时
Nebmode要改为1或2,因为它们已不再是反应物、产物或中间体,做能量最小化没有意义。取结构优化最后一步的两个端点结构,以及部分或全部中间像点结构(来自数据文件nebpath.xyz),作为初始结构,重新做CI-NEB优化。输入文件如下。 在本例中,两个端点的结构已经接近收敛( F_tang 和 F_perp 都非常小),为了提高计算速度,结构保持冻结(
Nebmode=2)。
$compass
basis-block
def2-SVP
end basis
geometry
# geom of image 1 (endpoint 1)
C 0.0094403 -0.0045178 0.0000000
N 0.0051489 -1.1595576 0.0000000
H 0.0054108 1.0740754 0.0000000
end geometry
norotate
$end
$bdfopt
solver
0
iopt
3
Trust
0.02
neb-block
crude
nebmode
2
nimage
3
end neb
nframe
4
geometry2
# geom of image 2
C 0.2822075 -0.0905533 0.0000000
N -0.4636856 -0.9729686 0.0000000
H 0.2014782 0.9735219 0.0000000
# geom of image 3
C 0.5703159 -0.2134900 0.0000000
N -0.5233808 -0.6609221 0.0000000
H -0.0269352 0.7844121 0.0000000
# geom of image 4
C 0.7857794 -0.5068651 0.0000000
N -0.3940425 -0.3160241 0.0000000
H -0.3717369 0.7328892 0.0000000
# geom of image 5 (endpoint 2)
C 0.5798873 -0.9890158 0.0000000
N -0.0251869 0.0162146 0.0000000
H -0.5347005 0.8828012 0.0000000
end geometry2
$end
$xuanyuan
$end
$scf
rks
dft
b3lyp
$end
$resp
geom
$end
这里用了一个小技巧:自动生成的中间像点坐标中,z方向坐标可能非零,三原子分子情况下会导致 Norotate 关键词与对称性程序发生冲突,
因此前面不得不用 Nosymm 关闭对称性。现在既然已经有了定性正确的像点坐标,我们可以把这些坐标在z方向的小值清零,
再把 Nosymm 改为 Norotate ,这样就可以利用Cs对称性加快计算。
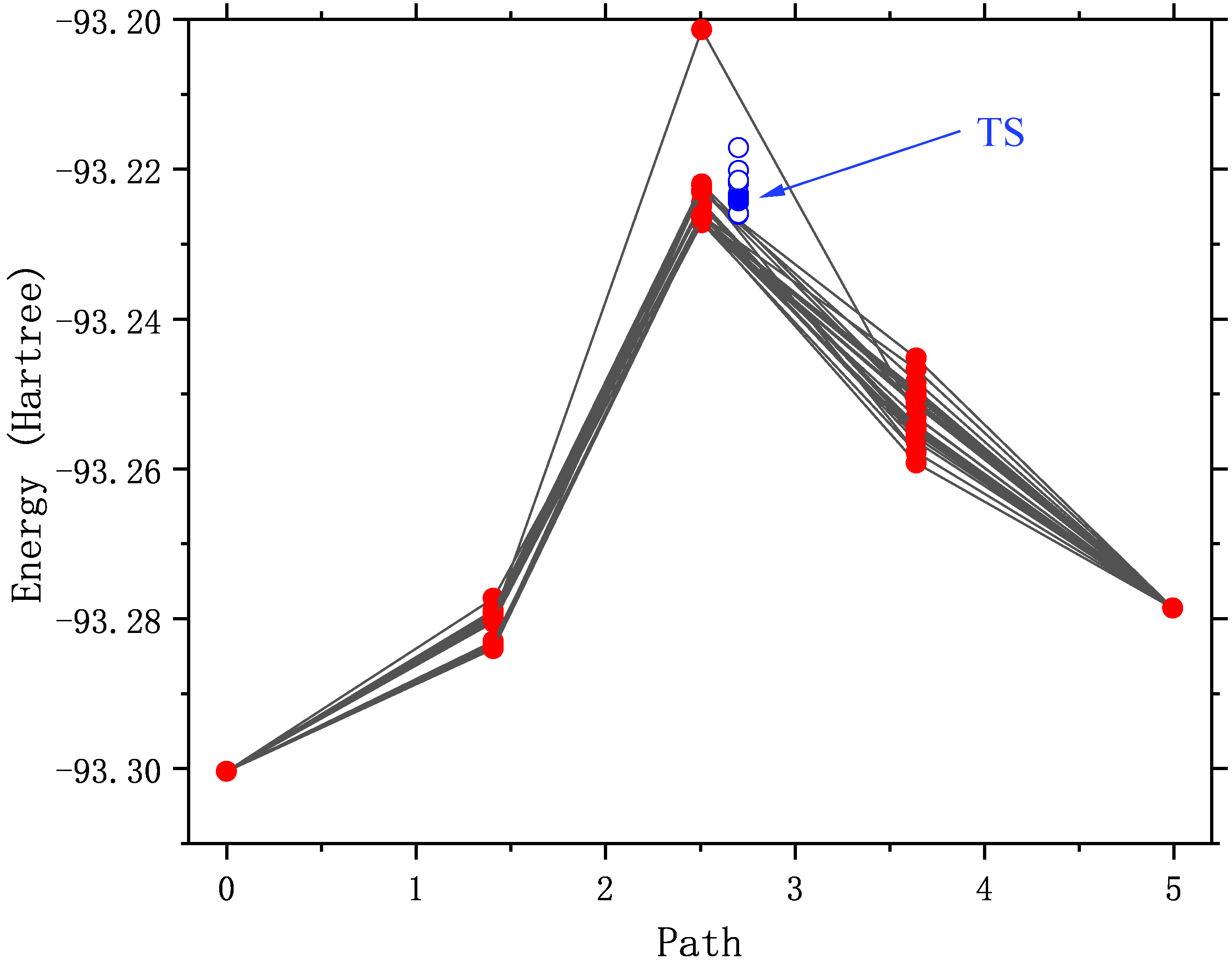
重新画NEB轨迹,如下图所示。由于中间像点使用了较好的初始结构,在优化前几步的能量已经合理了。
自旋混合态的结构优化:ZnS分子
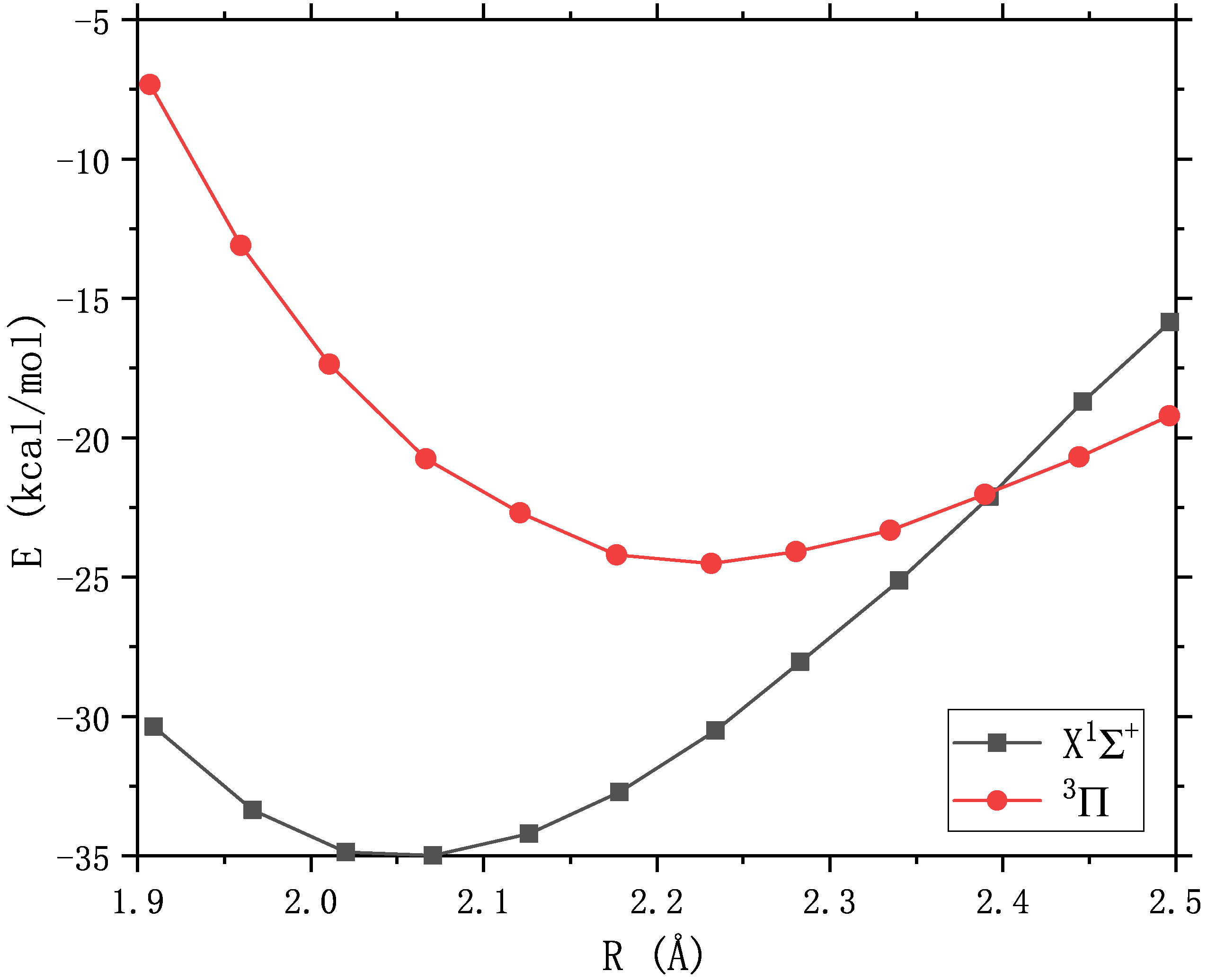
双原子分子ZnS的基态是闭壳层的 \(X^1\Sigma^+\) ,键长2.05埃;在11 kcal/mol以上有第一激发态 \(^3\Pi\) , 与基态在2.4埃附近发生交叉 [85] 。激发态的 \(^3\Pi_{0+}\) 分量与基态存在相互作用, 可能影响基态的键长。
如果对精度要求不高,可以用Truhlar等人建议的自旋轨道模型哈密顿 [86] 模拟两个自旋态之间的旋轨耦合, 考虑 \(^3\Pi_{0+}\) 对基态的影响。 输入文件如下。其中,基态用RKS完成,最低三重激发态用UKS完成。输入文件中有以下几点需要注意:
备注
两态混合计算的选项是
2soc(类似有3soc、4soc等)。一般来说,这两个自旋态需要有 不同的自旋多重度 ,除非理论计算证明两个相同自旋的态之间也存在强SOC相互作用(例如氮族原子的 \(^2D_u\) 和 \(^2P_u\) )。这里设置的旋轨耦合常数经验值400 \(\rm cm^{-1}\) 恰好是程序默认值,因此可以省略不写。
通过
solver= 1指定BDF自带优化器。也可以用DL-Find优化器,但是一般会慢一些。两个自旋态的能量、梯度要分别保存到
$BDFTASK.egrad.1和$BDFTASK.egrad.2文件。 至于哪个态规定为1号或2号,完全由用户决定,不影响最终结果。在BDF结构优化过程中,默认用上一步保存的SCF轨道作为当前SCF步骤的初猜,以获得最快的SCF收敛。由于两个SCF计算使用的SCF初猜轨道不同, 需要把它们分别备份为
$BDFTASK.scforb.1和$BDFTASK.scforb.2。然而当首次用它们覆盖$BDFTASK.scforb时,由于尚未进行SCF计算, 这两个文件不存在导致复制出错,BDF发现后会停止计算。为了屏蔽掉复制出错的信息,需要在复制命令的末尾加上2>/dev/null || :。最低三重激发态也可以用TDDFT通过自旋翻转计算,需要在
$tddft和$resp中加入一些额外的关键词(参见 TDDFT相关章节 )。 不过TDDFT不能很好描述电荷转移态(ZnS就属于这种情况),这里不采用。
$COMPASS
Title
two-state calculation of ZnS
Basis
lanl2dz
Geometry
Zn 0.0 0.0 0.0
S 0.0 0.0 2.05
END geometry
$END
$bdfopt
solver
1
multistate
2soc 400
$end
$xuanyuan
$end
%cp $BDF_WORKDIR/$BDFTASK.scforb.1 $BDF_WORKDIR/$BDFTASK.scforb 2>/dev/null || :
$SCF
rks
dft
pbe0
charge
0
spinmulti
1
$END
%cp $BDF_WORKDIR/$BDFTASK.scforb $BDF_WORKDIR/$BDFTASK.scforb.1
$resp
geom
$end
%cp $BDF_WORKDIR/$BDFTASK.egrad1 $BDF_WORKDIR/$BDFTASK.egrad.1
%cp $BDF_WORKDIR/$BDFTASK.scforb.2 $BDF_WORKDIR/$BDFTASK.scforb 2>/dev/null || :
$SCF
uks
dft
pbe0
charge
0
spinmulti
3
$END
%cp $BDF_WORKDIR/$BDFTASK.scforb $BDF_WORKDIR/$BDFTASK.scforb.2
$resp
geom
$end
%cp $BDF_WORKDIR/$BDFTASK.egrad1 $BDF_WORKDIR/$BDFTASK.egrad.2
计算结束后,优化的自旋混合基态键长是2.1485埃,比纯单重基态的键长2.1480埃(这个值远远大于高精度理论值2.05埃,是因为本算例采用的基组太小)略长一些, 说明 \(^3\Pi_{0+}\) 通过旋轨耦合作用把基态的键长拉长。最后一步优化的输出信息显示, \(^3\Pi_{0+}\) 在自旋混合基态中占了0.2%。
Multi-state calculation
-----------------------
Mixed-spin states by 2 scalar states.
Chi = 400.0 cm^-1 is used to constuct the SO model Hamiltonian.
/tmp/zouwl/BDF-1/test.egrad.1
E= -75.49718339
/tmp/zouwl/BDF-1/test.egrad.2
E= -75.46038704
Energies and weights of mixed-spin states:
------------------------------------------------------------------
No. E(mix) Weights
------------------------------------------------------------------
1 -75.49727344 99.8% 0.2%
2 -75.46029699 0.2% 99.8%
------------------------------------------------------------------
除了优化自旋混合基态的结构,还可以计算它的振动频率,为此需要把两个自旋态的Hessian分别保存到 $BDFTASK.hess.1 和 $BDFTASK.hess.2 文件。
如果仅在优化的结构上计算频率,和上面结构优化的输入类似,只需要把
solver= 1 改为hess=only,并把备份梯度文件$BDFTASK.egrad1改为备份Hessian文件$BDFTASK.hess。在做结构优化+频率计算时,输入要做额外修改。这是因为优化过程中不存在Hessian文件,而频率计算过程中不存在梯度文件,必须用
2>/dev/null || :屏蔽掉复制出错的信息。能够正常运行的输入文件如下:
$COMPASS
Title
two-state calculation of ZnS
Basis
lanl2dz
Geometry
Zn 0.0 0.0 0.0
S 0.0 0.0 2.05
END geometry
$END
$bdfopt
solver
1
multistate
2soc 400
hess
final
$end
$xuanyuan
$end
%cp $BDF_WORKDIR/$BDFTASK.scforb.1 $BDF_WORKDIR/$BDFTASK.scforb 2>/dev/null || :
$SCF
rks
dft
pbe0
charge
0
spinmulti
1
$END
%cp $BDF_WORKDIR/$BDFTASK.scforb $BDF_WORKDIR/$BDFTASK.scforb.1
$resp
geom
$end
%cp $BDF_WORKDIR/$BDFTASK.egrad1 $BDF_WORKDIR/$BDFTASK.egrad.1 2>/dev/null || :
%cp $BDF_WORKDIR/$BDFTASK.hess $BDF_WORKDIR/$BDFTASK.hess.1 2>/dev/null || :
%cp $BDF_WORKDIR/$BDFTASK.scforb.2 $BDF_WORKDIR/$BDFTASK.scforb 2>/dev/null || :
$SCF
uks
dft
pbe0
charge
0
spinmulti
3
$END
%cp $BDF_WORKDIR/$BDFTASK.scforb $BDF_WORKDIR/$BDFTASK.scforb.2
$resp
geom
$end
%cp $BDF_WORKDIR/$BDFTASK.egrad1 $BDF_WORKDIR/$BDFTASK.egrad.2 2>/dev/null || :
%cp $BDF_WORKDIR/$BDFTASK.hess $BDF_WORKDIR/$BDFTASK.hess.2 2>/dev/null || :
多态混合模型的主要用途是研究多态反应,优化自旋混合态的反应物,中间体,产物,过渡态, 以及反应路径。相比于MECP优化,它能提供更多信息(例如,如果MECP导致新的过渡态,多态混合模型可以估算MECP附近的振动频率和热化学量)。 当原子不太重的情况下(5d之前),多态混合模型比严格考虑旋轨耦合的二分量或四分量相对论方法有更多优势。
限制性结构优化
BDF还支持在结构优化中限制一个或多个内坐标的值,方法是在BDFOPT模块中加入constrain关键词。constrain关键词后的第一行为一个整数(以下称为N),表示总的限制数目;第2行到第N+1行定义每个限制。例如以下输入表示在结构优化时限制第2个原子和第5个原子之间的距离(这两个原子之间不一定需要有化学键):
$bdfopt
solver
1
constrain
1
2 5
$end
以下输入表示在结构优化时限制第1个原子和第2个原子之间的距离,同时还限制第2、第5、第10个原子形成的键角(同样地,不要求第2、第5个原子,或第5、第10个原子之间有化学键):
$bdfopt
solver
1
constrain
2
1 2
2 5 10
$end
以下输入表示在结构优化时限制第5、第10、第15、第20个原子之间的二面角,同时还限制第10、第15、第20、第25个原子之间的二面角:
$bdfopt
solver
1
constrain
2
5 10 15 20
10 15 20 25
$end
备注
即使分子坐标是以直角坐标而非内坐标的形式输入的,BDF仍然可以对内坐标做限制性优化。
当需要同时冻结多个原子(例如用团簇模型研究表面催化反应,欲冻结一部分催化剂原子)时,虽仍然可以穷尽这些原子之间的所有键长、键角、二面角并一一冻结,但是当需要冻结的原子数较多时,这样做比较麻烦、易错。因此BDF还支持冻结原子的笛卡尔坐标的功能,方法是使用frozen关键字。例如:
$bdfopt
...
frozen
3 # number of atoms to be frozen
2 -1
5 -1
10 -1
$end
表示冻结第2、5、10号原子。当使用DL-Find优化器(即solver=0)时,还可将-1替换为其他的数字,以只冻结该原子x、y、z坐标中的一个或两个:
0: free (default)
-1: frozen
-2: x frozen only
-3: y frozen only
-4: z frozen only
-23: x and y frozen
-24: x and z frozen
-34: y and z frozen
例如
$bdfopt
...
frozen
4 # number of atoms to be frozen
3 -1
4 -1
5 -1
6 -4
$end
即为冻结3~5号原子的笛卡尔坐标,以及6号原子的z坐标,但6号原子在x、y方向上仍可自由移动。
备注
(1)程序冻结的是用户指定的各原子之间的相对笛卡尔坐标,原子的绝对笛卡尔坐标仍可能因为分子标准取向的变化而变化; (2)同一个计算可以既冻结任意多的笛卡尔坐标,也冻结任意多的内坐标。
对于冻结内坐标的计算,程序还支持将内坐标冻结为某个和初始结构不同的值。例如以下输入文件,在M06-2X/6-31+G(d,p)/SMD(water)级别下优化环氧乙烷的结构,但限制其中一根C-O键长为2.0埃:
$compass
Geometry
C 0.00000000 0.70678098 -0.40492819
C 0.00000000 -0.70678098 -0.40492819
O 0.00000000 0.00000000 0.95133348
H 0.86041653 1.24388179 -0.74567167
H -0.86041653 1.24388179 -0.74567167
H 0.86041653 -1.24388179 -0.74567167
H -0.86041653 -1.24388179 -0.74567167
End Geometry
Basis
6-31+G(d,p)
MPEC+COSX
$end
$bdfopt
Solver
1
constraint
1 # number of constraints
2 3 = 2.0 # constrain the distance of atom 2 and atom 3 at 2.0 Angstrom
$end
$xuanyuan
$end
$scf
RKS
dft
M062X
solmodel
SMD
solvent
water
$end
$resp
geom
$end
初始结构中,两根C-O键的键长均为1.53埃,但优化收敛后,可以发现被冻结的C-O键的键长刚好为2.00埃(而不是1.53埃),另一根没有冻结的C-O键长则被优化为1.43埃:
|******************************************************************************|
Redundant internal coordinates on Angstrom/Degree
Name Definition Value Constraint
R1 1 2 1.4907 No
R2 1 3 1.4332 No
R3 1 4 1.0917 No
R4 1 5 1.0917 No
R5 2 3 2.0000 Yes
R6 2 6 1.0845 No
R7 2 7 1.0845 No
A1 2 1 3 86.29 No
...
同一个计算冻结的坐标中,可以只给一部分赋值,其余坐标自动沿用初始结构中的值。例如以下输入是合法的,表示冻结原子1、2、5的笛卡尔坐标,且冻结原子5-原子6-原子8的键角,以及原子11-原子12-原子13-原子14的二面角。其中除了原子5-原子6-原子8的键角被冻结为60度以外,其他几个被冻结的坐标的取值均与用户给定的初始结构一致,例如假如初始结构中原子11-原子12-原子13-原子14的二面角为120度,则在之后的结构优化中原子11-原子12-原子13-原子14的二面角始终为120度:
$bdfopt
...
solver
1
frozen
3
1 -1
2 -1
5 -1
constrain
2
5 6 8 = 60.
11 12 13 14
$end
备注
程序只支持设定被冻结的键长、键角、二面角值,而不支持在冻结笛卡尔坐标时设定被冻结的原子的笛卡尔坐标值。
当用户同时冻结多个内坐标并赋值时,用户需确认其冻结的内坐标值彼此自洽。作为内坐标不自洽的例子,考虑以下输入:
$bdfopt
Solver
1
constraint
3
1 2 = 1.0
1 3 = 2.0
2 3 = 4.0
$end
显然,这三个键长限制条件不能同时满足,因为无法由键长1埃、2埃、4埃的三根键构成一个三角形。程序发现无法同时满足三个限制条件后,打印警告信息:
q2c_coord_calc: warning: the 1-th constraint cannot be enforced, error = 0.204E+00
Check if the input constraints are mutually contradictory, or numerically ill-posed!
q2c_coord_calc: warning: the 2-th constraint cannot be enforced, error = 0.200E+00
Check if the input constraints are mutually contradictory, or numerically ill-posed!
q2c_coord_calc: warning: the 3-th constraint cannot be enforced, error = -0.149E+01
Check if the input constraints are mutually contradictory, or numerically ill-posed!
最后因多次重启优化器仍然无法解决该问题,程序报错退出。
备注
以上算例中,同时满足各个内坐标限制条件是数学上不可能的。有时虽然同时满足各个限制条件在数学上可能,但初始结构离满足限制条件的结构太远,此时程序仍然可能找不到同时满足所有限制条件的结构,并打印警告或报错退出。
柔性扫描
2025年6月及之后的BDF版本支持柔性扫描功能,可以做任意维的柔性扫描,且各个扫描点既可以是等间距的(网格扫描),也可以不等间距(散点扫描)。如以下写法表示将第5个原子与第6个原子之间的键长从4.0埃扫描至1.0埃,步长0.1埃:
$bdfopt
solver
1 # 柔性扫描必须指定solver=1
scan
1 # 扫描1个坐标
5 6 = 4.0 1.0 -0.1 # 注意因为从大到小扫描,步长是负的
$end
以下写法表示将第5个原子与第6个原子之间的键长从4.0埃扫描至1.0埃,但扫描步长并非定值,只扫描4.0、3.0、2.5、2.0、1.8、1.6、1.4、1.2、1.0埃共9个键长:
$bdfopt
solver
1
scan
1 9 # 扫描1个坐标,扫9个点
5 6 # 这个坐标是第5个原子与第6个原子之间的键长
4.0 # 从这行开始是键长的值
3.0
2.5
2.0
1.8
1.6
1.4
1.2
1.0
$end
以下写法表示做二维扫描,第一个维度是原子2、4、5的键角,第二个维度是原子2、4、5、13的二面角,前者从90度扫到120度,步长5度,后者从0度扫到360度,步长10度:
$bdfopt
solver
1 # 柔性扫描必须指定solver=1
scan
2 # 扫描2个坐标
2 4 5 = 90 120 5
2 4 5 13 = 0 360 10
$end
在上例中,程序首先固定键角为90度,将二面角从0度、10度……一直扫到360度(360度在程序里显示为0度,但因为扫描的每一步都是以前一步的结构作为初猜的,扫回0度以后的结构未必与扫描的第一个点的0度结构相符),然后才将键角增加为95度,继续将二面角从0度、10度……一直扫到360度,以此类推。
以下写法表示将原子1、2所成的键从1.5埃扫到2.0埃,步长0.1,并固定原子3、4所成的键的键长与原子1、2所成的键的键长相等:
$bdfopt
solver
1
scan
2 6 # 扫描2个坐标,扫6个点
1 2
3 4
1.5 1.5 # 从这行开始是键长的值
1.6 1.6
1.7 1.7
1.8 1.8
1.9 1.9
2.0 2.0
$end
由上例可以看出,对于多根键同时形成的协同反应的过渡态初步搜索,一维扫描无法胜任时,无需做完整的二维扫描(需要扫36个点),只需扫描二维势能面上较重要的区域,即两根键键长相同的区域(只需扫6个点),由此可以大大节省时间。若已知该反应并非严格的同步反应,即一根键形成得比另一根键稍早,也可以相应地调整需要扫描势能面上的哪些点,使一根键的键长随扫描过程增加得比另一根键更快。
扫描结果以三种形式输出。首先,可以从输出文件看到当前扫描的内坐标:
=================================================
Relaxed surface scan step 1
Bond 1 2 = 1.10000 Ang
Angle 1 2 3 = 180.00000 Deg
=================================================
每打印一次 Relaxed surface scan step ,都会打印若干步限制性优化的信息(即若干组结构、能量和梯度),其中只有最后一步才是收敛的柔性扫描曲线/曲面结果。
其次,可以从 $BDFTASK.scanpes 文件读取扫描结果汇总信息:
Scan step 1
Bond 1 2 = 1.10000000
Angle 1 2 3 = 180.00000000
Energy= -91.68864589
Geometry=
H 0.00000000 0.00000000 -1.59772393
C 0.00000000 0.00000000 -0.49772393
N 0.00000000 0.00000000 0.65486679
Gradient=
H 0.00000000 -0.00000000 -0.02545813
C -0.00000000 -0.00000000 0.02525156
N 0.00000000 0.00000000 0.00020656
Scan step 2
Bond 1 2 = 1.10000000
Angle 1 2 3 = 150.00000000
Energy= -91.67474826
Geometry=
H -1.54410087 -0.13580336 0.00000000
C -0.48560359 0.16350169 0.00000000
N 0.63681749 -0.12074382 0.00000000
Gradient=
H -0.01385239 -0.03003724 0.00000000
C 0.01979162 0.05316515 0.00000000
N -0.00593924 -0.02312791 0.00000000
...
最后,每步结构优化的结构会输出到 $BDFTASK.optgeom1 、 $BDFTASK.optgeom2 ……等文件中,其与一般的 .optgeom 文件格式相同,可进行可视化、格式转换、后续计算等。
激发态结构优化
BDF程序除优化基态结构外,还可优化激发态结构,具体请参见 TDDFT相关章节 ,此处不再赘述。
QM/MM结构优化
BDF还可以用QM/MM组合方法进行结构优化,但与纯QM结构优化不同的是,QM/MM结构优化不能使用BDFOPT模块完成,而必须借助pDynamo程序自带的结构优化函数。具体请参见 QM/MM相关章节 ,此处不再赘述。
自动消除虚频
不管是优化极小值点结构还是优化过渡态,都经常会遇到优化收敛的结构的虚频数目与预期不符的问题,具体可分为3类:(1)优化极小值点收敛的结构有虚频;(2)优化过渡态收敛的结构的虚频多于1个;(3)优化过渡态收敛的结构没有虚频。对于(1)、(2)两类情况,BDF可以自动消除多余的虚频,为此需要在BDFOPT模块的输入里添加 rmimag (或 removeimag )关键词;该关键词对于(3)的情况也有一定作用,即在优化过渡态结果没有虚频时可以在附近寻找有一个虚频的结构,但成功率较低。例如以下输入表示,优化极小值点结构,然后做一个频率计算,如果没有虚频则结束计算;如果有虚频则自动将分子结构沿着绝对值最大的虚频对应的振动模的方向扰动,然后继续优化,优化收敛后再做一次频率计算验证虚频有没有消掉,如此反复直至所有虚频完全消掉,或计算了10次频率仍然没办法消掉所有虚频为止:
$bdfopt
solver
1
rmimag
$end
以下输入和上述输入的效果类似,区别在于会对最后一次算出的Hessian做完整的频率分析和热化学分析:
$bdfopt
solver
1
rmimag
hess
final
$end
以下输入表示,优化过渡态结构(在优化开始前先做一个频率计算,来提供结构优化的初始Hessian),然后做一个频率计算,如果恰有1个虚频则结束计算。如果虚频数目大于1,则自动将分子结构沿着绝对值第二大的虚频的方向扰动,然后继续优化,优化收敛后再做个频率计算验证多余的虚频有没有消掉,如此反复直至虚频数目等于1。如果虚频数目等于0,则自动尝试在附近寻找虚频数目等于1的结构,优化收敛后同样重新计算频率,验证虚频数目,如此反复直至虚频数目等于1:
$bdfopt
solver
1
rmimag
hess
init # calculate initial Hessian. If a thermochemistry analysis on the final Hessian is desired, change “init” to “init+final”
iopt
10 # transition state optimization
$end
以下是一个完整的算例,该算例在HFLYP/6-31G(d)水平下优化了 \(\ce{ClF3}\) 的平衡结构:
$compass
title
ClF3
basis
6-31G(d)
geometry
Cl 0.000000 0.000000 0.000000
F -2.966870 0.000000 0.000000
F 1.483435 2.569385 0.000000
F 1.483435 -2.569385 0.000000
end geometry
unit
bohr
nosymm
$end
$bdfopt
solver
1
rmimag
hess
final
$end
$xuanyuan
$end
$scf
rks
dft
HFLYP
$end
$resp
geom
$end
初始结构符合 \(\rm D_{3h}\) 对称性,但因为预期优化收敛的结构可能存在虚频,在 compass 模块里用 nosymm 关键字指定在 \(\rm C_{1}\) 群下计算。程序首先收敛到 \(\rm D_{3h}\) 点群下的极小值点:
|******************************************************************************|
Redundant internal coordinates on Angstrom/Degree
Name Definition Value Constraint
R1 1 2 1.6666 No
R2 1 3 1.6667 No
R3 1 4 1.6667 No
A1 2 1 3 119.99 No
A2 2 1 4 119.99 No
A3 3 1 4 120.02 No
D1 4 1 3 2 179.97 No
D2 2 1 4 3 179.97 No
D3 3 1 4 2 -179.97 No
|******************************************************************************|
接着重启结构优化器:
--- Restarting optimizer ... (10 attempt(s) remaining) ---
接下来程序会进行一个数值Hessian计算,发现该结构有两个虚频:
Warning: the number of imaginary frequencies, 2, is different from the desired number, 0!
因此程序对该结构进行扰动,并继续优化,以期消除虚频。其中因一个F-Cl-F键角接近180°,需要重新构建冗余内坐标,导致结构优化器再次重启。最后收敛得到属于 \(\rm C_{2v}\) 点群、呈T字形的结构:
|******************************************************************************|
Redundant internal coordinates on Angstrom/Degree
Name Definition Value Constraint
R1 1 2 1.5587 No
R2 1 3 1.6470 No
R3 1 4 1.6470 No
R4 2 4 2.1859 No
A1 2 1 3 85.95 No
A2 2 1 4 85.94 No
|******************************************************************************|
最后第3次重启结构优化器,经过Hessian计算确认,该结构没有虚频,即一开始的两个虚频已经消除。此时整个计算宣告收敛:
*************************************** Properties of Normal Modes ***************************************
Results of vibrations:
Normal frequencies (cm^-1), reduced masses (AMU), force constants (mDyn/A)
1 2 3
Irreps A" A' A'
Frequencies 385.8687 414.4702 519.9076
Reduced masses 24.3196 21.5030 19.4352
Force constants 2.1335 2.1764 3.0952
备注
(1)程序不能保证在所有情况下都能消除所有多余的虚频,即便程序正常结束,虚频的数目也仍然可能是错误的。所以即便加了 rmimag 关键字,优化结束后用户仍然需要检查虚频数目。如果虚频数目仍然不等于预期值(也就是对于极小值点优化,仍然有虚频;或者对于过渡态优化,没有虚频或者虚频数目大于1个),则需要按 虚频问题 小节的方法手动处理。
(2)如果分子具有点群对称性,但计算时没有指定 nosymm ,则可能无法完全消除所有的虚频,甚至可能导致结构优化不收敛。例如在以上算例中,如果不指定 nosymm ,即令计算在分子的实际点群( \(\rm D_{3h}\) )下进行,则因为不管如何消除两个虚频都会破坏 \(\rm D_{3h}\) 对称性,使得无法消除虚频,进而导致优化不收敛。
(3)限制性优化(不管限制的是笛卡尔坐标还是内坐标)得到的结构,有可能(但不一定)存在无法消掉的虚频。此时如果虚频的数目大于预期值,未必说明当前结构不可用。用户应通过观察虚频的振动模式,自行判断虚频是否是优化时施加的限制条件所导致的,进而判断是否应当消除这些虚频。
锥形交叉点(CI)和最低能量交叉点(MECP)的优化
优化CI和MECP需要调用DL-FIND外部库 [84] ,为此需要在BDFOPT模块的输入里添加以下关键词
solver
0
相应地,前述各算例的 solver 1 代表使用BDF自带的结构优化代码而非DL-FIND来进行优化。原则上,极小值点和过渡态的优化也可用DL-FIND来实现,但效率一般不如BDF自带代码好,因此仅对于CI、MECP优化等BDF自带代码不支持的任务,才应调用DL-FIND。
以下为CI优化的示例输入,该输入文件计算了乙烯的T1态和T2态的锥形交叉点:
#----------------------------------------------------------------------
# Gradient projection method for CI between T1 and T2 by TDDFT BHHLYP/6-31G
#
$COMPASS
Title
C2H4 Molecule test run
Basis
6-31G
Geometry
C 0.00107880 -0.00318153 1.43425054
C 0.00066030 0.00195132 -1.43437339
H 0.05960990 -0.89114967 0.84012371
H -0.05830329 0.95445870 0.96064844
H 0.05950228 0.89180839 -0.84311032
H -0.06267534 -0.95390169 -0.95768311
END geometry
nosymm
$END
$bdfopt
imulti #优化CI
2
maxcycle #最大优化步数
50
tolgrad #均方根梯度的收敛标准
1.d-4
tolstep #均方根步长的收敛标准
5.d-3
$end
$xuanyuan
$end
$SCF
RKS
charge
0
spinmulti
1
atomorb
DFT
BHHLYP
$END
$tddft
imethod
1
isf
1
itda
1
nroot
5
idiag
1
istore
1
crit_e
1.d-8
crit_vec
1.d-6
lefteig
ialda
4
$end
$resp
geom
norder
1
method
2
iroot
1
nfiles
1
$end
$resp
geom
norder
1
method
2
iroot
2
nfiles
1
$end
$resp
iprt
1
QUAD
FNAC
double
norder
1
method
2
nfiles
1
pairs
1
1 1 1 1 1 2
$end
注意该任务不仅需要计算T1态和T2态的梯度,还需要计算T1态和T2态之间的非绝热耦合矢量(由最后一个RESP模块完成),相关关键词参见 tddft ,此处不再赘述。在BDFOPT模块的输入中, imulti 2 代表优化CI。和普通结构优化任务类似,CI优化会输出每步的梯度和步长收敛情况,与此同时还会输出能量收敛情况。例如以上算例最后一步优化的输出为:
Testing convergence in cycle 6
Energy 0.0000E+00 Target: 1.0000E-06 converged? yes
Max step 9.0855E-04 Target: 5.0000E-03 converged? yes component 4
RMS step 5.6602E-04 Target: 3.3333E-03 converged? yes
Max grad 5.5511E-05 Target: 1.0000E-04 converged? yes component 1
RMS grad 2.7645E-05 Target: 6.6667E-05 converged? yes
Converged!
converged
与前述各类优化任务类似,收敛的CI结构保存于 .optgeom 文件内,坐标单位为Bohr。注意能量一行的值总是显示为0,这并不代表CI优化时体系能量不变,而是因为优化CI不会用到能量的收敛情况来判断是否收敛。出于同样的原因, tolene 关键词对于CI优化(以及下述的MECP优化)是没有作用的。
以下是优化MECP的示例输入文件:
#----------------------------------------------------------------------
# Gradient projection method for MECP between S0 and T1 by BHHLYP/6-31G
#
$COMPASS
Title
C2H4 Molecule test run
Basis
6-31G
Geometry
C -0.00000141 0.00000353 0.72393424
C 0.00000417 -0.00000109 -0.72393515
H 0.73780975 -0.54421247 1.29907106
H -0.73778145 0.54421417 1.29907329
H 0.73777374 0.54421576 -1.29907129
H -0.73779427 -0.54423609 -1.29906321
END geometry
nosymm
$END
$bdfopt
imulti
2
maxcycle
50
tolgrad
1.d-4
tolstep
5.d-3
noncouple
$end
$xuanyuan
$end
$SCF
RKS
charge
0
spinmulti
1
atomorb
DFT
BHHLYP
$END
$resp
geom
norder
1
method
1
$end
$SCF
UKS
charge
0
spinmulti
3
atomorb
DFT
BHHLYP
$END
$resp
geom
norder
1
method
1
$end
其中 imulti 2 和 noncouple 关键词指定进行MECP优化。注意MECP优化任务仅需计算两个态(此处为S0态和T1态)的梯度,而无需计算非绝热耦合矢量。MECP优化任务的输出与CI优化任务类似,此处不再赘述。
几何优化常见问题
虚频问题
几何结构优化不仅要求结构收敛(即梯度和步长满足收敛限要求),同时还要求所得结构的虚频数目符合预期值,即当优化极小值点结构时,虚频数目为0;优化过渡态时,虚频数目为1;若虚频数目大于1为高阶鞍点。当实际计算得到的虚频数目与预期值不符时,需要调整结构并重新优化。一般情况下,利用 rmimag关键字 可以解决大部分的虚频数目过多的问题,以及一小部分虚频数目过少的问题。当 rmimag 关键字无法奏效时,用户应当按以下方法手动解决虚频数目不符合预期的问题:
当实际计算得到的虚频数目小于预期值,也即优化过渡态得到虚频数量为0的结构时:此时一般说明得到的过渡态结构定性错误,需要根据化学常识重新准备初猜结构。
当实际计算得到的虚频数目大于预期值时,此时存在两种可能情况:(1)虚频是因为计算的数值误差所导致的,并非真实存在。此时可以通过加大格点、减小积分截断阈值、减小各类收敛阈值(如SCF收敛阈值、结构优化收敛阈值等)等方法解决。(2)体系确实存在虚频。此时应当从输出文件查看虚频对应的简正模,并沿着该简正模方向对收敛的结构进行扰动,然后以扰动后的结构为初猜结构,重新进行优化。
注意无法仅从频率计算结果判断某个虚频是否是数值误差导致的,但一般而言,虚频的绝对值越小,就越可能是数值误差导致的,反之则越可能是真实存在的。
限制性优化(不管限制的是笛卡尔坐标还是内坐标)得到的结构,有可能(但不一定)存在无法消掉的虚频。此时如果虚频的数目大于预期值,未必说明当前结构不可用。用户应通过观察虚频的振动模式,自行判断虚频是否是优化时施加的限制条件所导致的,进而判断是否应当消除这些虚频。
对称性问题
当初始结构具有 \(\rm C_1\) 群以上的点群对称性时,结构优化有可能会破坏点群对称性,例如优化氨分子,初始结构对称性为 \(\rm D_{3h}\) 的平面结构时,结构优化可能会得到对称性为 \(\rm C_{3v}\) 的锥形结构。 默认情况下BDF会强制保持分子点群对称性,除非体系存在一阶Jahn-Teller效应。如果用户希望BDF破坏分子的对称性,可以采取以下方法之一:
仍然在高对称性下优化至收敛,然后计算频率。若存在虚频,按照上一小节方法扰动分子结构来消除虚频。如果分子可以通过破坏对称性来进一步降低能量,那么此时应该发现扰动后的分子结构的对称性已有所降低,继续以该结构为初始结构进行优化即可。
在COMPASS模块中指定采用分子点群的某一个子群,此时程序只会保持该子群对称性不被破坏。若指定的是 \(\rm C_1\) 群,则程序允许以任何方式破坏分子对称性,可以最大程度上提高得到低能量结构的概率,但代价为无法利用点群对称性加速计算,导致计算量增加。
几何优化不收敛
导致几何优化不收敛的因素有很多,包括:
能量、梯度存在数值噪声,往往表现为置信半径不断减小,步长收敛或接近收敛,但梯度离收敛较远;
势能面过于平缓;
分子有不止一个稳定波函数,结构优化时波函数在各个稳定解之间来回跳跃,不能稳定地始终收敛到同一个解;
分子结构不合理,如坐标单位错误(如坐标的单位本来是Bohr,但输入文件里指定的单位是Angstrom,或反之),多画或漏画原子,非成键原子之间的距离太近,等等。
某些反应不存在过渡态(要么正反应的活化能为0,要么逆反应的活化能为0),用户如果误以为存在过渡态,进行过渡态优化,则优化不收敛或总是收敛到错误的结构。因此,当过渡态优化始终不收敛时,用户应当考虑当前反应本来就不存在过渡态的可能性,例如做一个柔性扫描,看扫描曲线是否单调变化,如果在整个反应过程中扫描曲线均为单调、平滑变化,则可以认为不存在过渡态(前提是扫描的键长/键角/二面角范围足够大,且扫描步长足够小)。
某些激发态结构优化可能会优化到锥形交叉点附近。例如优化S1结构,有时会优化到S0/S1锥形交叉点附近,此时因为锥形交叉点是附近唯一的局部极小值点,但是势能面在该处不可导,无法满足梯度为0的条件,因此结构优化会在锥形交叉点附近无限振荡。此时用户应重新评估优化S1平衡结构的目的,例如如果是为了计算荧光发射,且从S1势能面上的Franck-Condon点可以直接优化到S0/S1锥形交叉点,则可以较为确定地认为该体系的S1态不发荧光,因为S1态极易从S0/S1锥形交叉点发生内转换弛豫到S0态。必要时,用户可改用 锥形交叉点的优化算法 来优化得到S0/S1锥形交叉点,而不受该点处势能面不可导的影响。
某些TDDFT结构优化可能会优化到势能面上基态波函数不稳定的区域(例如某根键显著拉长,产生双自由基性质),此时程序会因TDDFT出现虚激发能(极个别时候为复激发能),而在RESP模块报错退出。此时用户应根据自己的计算目的,试图收敛得到稳定波函数后再进行计算,或改用TDA,或计算某个更高的激发态。注意这几种方法的结果是不等价的,用户应明确自己的计算目的后再做选择,不可仅因其中一种方法解决了虚激发能的问题,就盲目相信其结果。
极个别情况下,因为程序构建的内坐标含有接近180度的键角,导致优化失败。如前所述,一般来说程序会反复尝试重新构建内坐标,直至尝试10次仍未收敛才报错退出,所以BDF因内坐标问题报错退出的概率应较其他很多量化程序低。
如遇到几何优化不收敛,或虽然尚未达到最大收敛次数但毫无收敛趋势的情形,经反复检查分子三维结构无误且合理(所谓结构合理,既包括用户提供的初始结构无误,例如没有漏画、多画、错画原子等,也包括优化后的结构合理,没有不符合化学常识的结构拥挤、扭曲等情况)、波函数收敛正常以后,可依次尝试以下各方法解决:
以优化不收敛的任务的最后一帧结构为初始结构,重新开始优化。除了手动将最后一帧的结构坐标复制到输入文件里以外,一个更简单的办法是在COMPASS模块里加入
restart关键词,如:
$compass
title
CH3Cl geomopt
basis
def2-SV(P)
geometry
C 2.67184328 0.03549756 -3.40353093
H 2.05038141 -0.21545378 -2.56943947
H 2.80438882 1.09651909 -3.44309081
H 3.62454948 -0.43911916 -3.29403269
Cl 1.90897396 -0.51627638 -4.89053325
end geometry
restart
$end
假设输入文件的文件名为 CH3Cl-opt.inp ,则此时程序自动读取 CH3Cl-opt.optgeom 里的坐标作为初始结构(注意此时程序虽然不会用到 geometry 字段里的分子坐标,但该分子坐标不能删去)。乍看起来,这样做似乎与简单地增加几何优化最大迭代步数无异,但实际上这样做的效果往往比单纯增加最大迭代步数更好,例如优化100步后重新读取结构再优化50步,收敛概率常常比连续迭代150步更高,这是因为重新读取结构继续优化时,程序重新产生了初始Hessian,进而避免了准牛顿法连续多步近似构建Hessian所累积的误差。该方法也适用于虽然结构优化器没有报错,但SCF/TDDFT等不收敛导致报错的情形。
需要注意的是,不是所有情况下都可以只在输入文件里添加restart就可以重新运行,某些情况下需要改变输入文件的其他信息。例如:(1)若结构优化改变了点群对称性,可能需要相应地改变点群名字、轨道/激发态不可约表示相关设置等。(2)对于使用了态跟踪的TDDFT计算,需要重新设置resp模块的iroot关键字。如某个结构优化任务优化第3个激发态,随后态跟踪算法切换为跟踪第2个态,然后计算因为不收敛等问题而中断了,此时重新提交计算时,除添加restart关键词以外,还需将resp模块的iroot关键字从-3改为-2。(3)因结构优化不收敛以外的原因终止的任务,建议添加与解决问题有关的关键字,如SCF不收敛终止的结构优化任务,建议添加帮助SCF收敛的关键字,再提交计算。
减小优化步长,或称置信半径(trust radius)。方法为使用trust关键词,如
$bdfopt
solver
1
trust
0.05
$end
默认的置信半径为0.3,因此新设置的置信半径应当小于0.3。注意程序如果检测到置信半径太小,会动态地增加置信半径,为了避免这一行为,可以将置信半径设为负值,如
$bdfopt
solver
1
trust
-0.05
$end
即表示,初始置信半径设为0.05,且在整个结构优化过程中禁止置信半径超过0.05。
对于过渡态优化,可用
recalchess关键词指定每隔若干步重新计算精确Hessian。如
$bdfopt
solver
1
iopt
10
hess
init
recalchess
10
$end
表示除在结构优化之前计算精确Hessian外,每隔10步结构优化重新计算一次精确Hessian。
加大格点,减小积分截断阈值及SCF等的收敛阈值,以减小数值误差。注意该方法只在结构优化几乎收敛但无法完全收敛时有用。
改用DL-Find优化器,在直角坐标下优化结构:
$bdfopt
solver
0
$end
其中 solver 0 表示使用DL-Find优化器而非BDF自带的优化器。因DL-Find默认在直角坐标下优化结构,所以无需用额外的关键词指定在直角坐标下优化。该方法适用于程序因内坐标键角接近180度,经多次重启优化器仍无法解决进而报错的情形(此时应跳过以上各解决方法,直接尝试本方法,因为这种情况下本方法解决问题的概率最大,如不能解决问题再依次尝试以上各方法),也适用于虽然程序报错不是因为键角接近180度问题,但所计算体系不太适合用内坐标描述,因而优化不收敛的情形(例如体系是较大的非共价团簇)。
在应用以上方法之前,用户应检查当前这个不收敛的计算所得结构相比用户提供的初猜结构哪个更合理,用其中较为合理的那个结构作为接下来重新优化的初猜结构。如果不仅结构优化后的结构不合理,还发现初猜结构也不合理,则应重新准备初猜结构。这对于过渡态优化尤其重要,如果第一次结构优化时分子结构被优化到过渡态区域以外,则之后无论尝试什么方法也很难将结构重新优化回过渡态区域附近。因此,不经检查结构是否合理、盲目地以前一次结构优化的最后一帧结构(或前一次结构优化的初猜结构)作为下一次优化的初猜结构,是不可取的。