溶剂化模型
溶剂化模型用于计算溶质和溶剂之间的相互作用,一般分为隐式溶剂模型(连续介质模型)和显式溶剂模型两种。 在BDF中,对于连续溶剂模型,可以选择IEFPCM、SS(V)PE、CPCM、COSMO、 ddCOSMO(domain-decomposition COSMO solvation model)以及SMD,对于显示溶剂模型采用QM/MM方法,结合pDymamo2.0程序包进行计算。
BDF溶剂化模型支持的功能:
PCMs |
Ground state |
Excited state |
|||
|---|---|---|---|---|---|
Single-point |
Gradient |
Hessian |
Single-point |
Gradient |
|
COSMO |
√ |
√ |
√ |
√ |
√ |
CPCM |
√ |
√ |
√ |
√ |
√ |
SS(V)PE |
√ |
√ |
√ |
√ |
√ |
IEFPCM |
√ |
√ |
√ |
√ |
√ |
SMD |
√ |
√ |
√ |
√ |
√ |
溶剂类型设置
在 SCF 模块中加入 solvent 关键词,表示要进行溶剂化效应计算,紧跟一行可以输入溶剂类型,比如 water 。
以甲醛分子在水溶液中的计算为例,其输入文件为:
$COMPASS
Title
ch2o Molecule test run
Basis
6-31g
Geometry
C 0.00000000 0.00000000 -0.54200000
O 0.00000000 0.00000000 0.67700000
H 0.00000000 0.93500000 -1.08200000
H 0.00000000 -0.93500000 -1.08200000
END geometry
nosymm
unit
ang
$END
$xuanyuan
$END
$SCF
rks
dft
b3lyp
solvent #溶剂化计算开关
water #指定溶剂
grid
medium
$END
溶剂类型的指定可以输入 BDF支持的溶剂类型列表 中的名称或者别名。对于表中没有的溶剂,可以输入介电常数。格式如下:
solvent
user #用户指定
dielectric
78.3553 #输入介电常数
溶剂模型设置
连续介质模型是将溶剂视为有一定介电常数的可极化的连续介质。
目前BDF支持的溶剂模型有ddCOSMO、COSMO、CPCM、IEFPCM、SS(V)PE以及SMD, 对应的关键词为 ddcosmo 、 cosmo、 cpcm、 iefpcm、 ssvpe、 smd。输入为:
solvent
water
solmodel
IEFPCM #溶剂模型
未指定特定模型时,将默认使用IEFPCM模型。对于COSMO和CPCM,可以通过 cosmoFactorK 来指定the dielectric screening factor, \(f_\epsilon=\frac{\epsilon-1}{\epsilon+k}\) ,中k的大小。对于COSMO,k默认为0.5;对于CPCM,k默认为0。
cosmoFactorK
0.5
对于SMD模型,可以手动指定溶剂的折射率、Abraham氢键酸度、Abraham氢键碱度、表面张力、芳香度、卤素度
refractiveIndex # 折射率
1.43
HBondAcidity # Abraham氢键酸度
0.229
HBondBasicity # Abraham氢键碱度
0.265
SurfaceTensionAtInterface # 表面张力
61.24
CarbonAromaticity # 芳香度
0.12
ElectronegativeHalogenicity # 卤素度
0.24
备注
使用SMD模型将关闭 溶剂化自由能非静电部分 的计算,取而代之将计算SMx系列的 \(\Delta G_{CDS}\)
孔穴自定义设置
连续介质模型将视为连续介质的溶剂根据溶质分子来形成孔穴,孔穴的形状会对溶剂化能的计算产生较大的影响。对于连续介质模型,有多种孔穴的定义:vdW(van der Waals surface), SES(solvent-excluded surface), SAS(solvent-accessible surface)等。
在BDF中默认采用1.1倍的UFF半径来构建vdW表面的孔穴。
对于COSMO、CPCM、IEFPCM、SS(V)PE以及SMD溶剂模型,可以通过 cavity, RadiusType, vdWScale, radii, uatm, acidHRadius 等关键词来自定义孔穴的形状。
cavity # 生成孔穴表面的方式
swig # swig | switching | ses | sphere,默认为 swig
uatm # 联合原子拓扑方法
false # false | true,默认为 false
radiusType
UFF # UFF | Bondi,默认为 UFF
vdWScale
1.1 # 默认 1.1, 即 1.1 倍 RadiusType 半径
radii
1=1.4430 2=1.7500 # 第一个原子的半径设为 1.4430Å, 第二个原子的半径设为 1.7500Å
# 等号间不能有空格, 一行最多128字符, 一行写不下可以加上radii之后新增一行
# radii的设置会覆盖 vdWScale*RadiusType 中相同原子的设置
radii
H=1.4430 O=1.7500 # 同上, 将 H 原子的半径设为1.4430Å, 将 O 原子的半径设为 1.7500Å。两种方式可以混合使用。
acidHRadius # 单独设置酸性H半径,单位 Å
1.2
通过 cavity 关键词,可以控制生成孔穴表面的方式
switching表示用平滑函数来处理vdW表面的格点权重swig表示 switching/gaussian,即在switching的基础上再使用高斯函数对格点处的点电荷做平滑处理sphere表示形成一个圆球状的孔穴来包裹整个分子。
uatm 表示将H原子联合进重原子共同形成孔穴。
另外还可以通过 cavityNGrid 或 cavityPrecision 来指定孔穴的格点精度(每个原子表面的最大tesserae数)。
cavityNGrid # 控制每个原子生成的孔穴表面的格点数, 会自动调整至最近的 lebedev 格点
302 # 默认为 302
# 或者
cavityPrecision
medium # ultraCoarse | coarse | medium | fine | ultraFine,默认为 medium
基态溶剂化能计算
对于基态溶剂化能计算,通常只需要在 SCF 模块中设定溶剂类型以及溶剂化模型,其余参数使用默认即可。
以甲醛分子在水溶液中使用SMD模型的计算为例,其输入文件为:
$COMPASS
Title
ch2o Molecule test run
Basis
6-31g
Geometry
C 0.00000000 0.00000000 -0.54200000
O 0.00000000 0.00000000 0.67700000
H 0.00000000 0.93500000 -1.08200000
H 0.00000000 -0.93500000 -1.08200000
END geometry
$END
$xuanyuan
$END
$SCF
rks
dft
gb3lyp
solvent #溶剂化计算开关
water #指定溶剂
solmodel #指定溶剂化模型
smd
$END
备注
使用 cosmosave 关键词可以保存孔穴体积、表面积,tesserae坐标、电荷、面积等信息到工作目录的.cosmo文件,如有需要,可以利用 $BDFHOME/sbin/conv2gaucosmo.py 可以将其转化为类似gaussian的cosmors关键词所生成的文件的格式。
非静电溶剂化能计算
溶剂化自由能包括静电溶剂化能以及非静电溶剂化能。上述的PCM模型计算了静电溶剂化能。非静电溶剂化能一般可以分为为孔穴能 \(\Delta G_{cav}\) 和色散-排斥能 \(\Delta G_{dis-rep}\) 。 孔穴能是在假设溶质溶剂之间无相互作用时,将溶质分子从气相移入液相形成孔穴所做的功。可以用基于定标粒子理论(SPT)的Pierotti-Claverie公式来进行计算。色散能与排斥能可以用粒子对势近似法来计算。
在BDF中,默认不开启非静电溶剂化能的计算,可以通过以下关键词来开启非静电溶剂化能的计算
nonels
dis rep cav # 色散能 排斥能 孔穴能
solventAtoms # 溶剂分子的各类型原子的个数(分子式)
H2O1 # 默认为H2O1,不能省略1,因为不区分大小写后无法确定元素符号是几个字母
solventRho # 溶剂分子数密度,单位 molecules Å^-3
0.03333
solventRadius # 溶剂分子半径,单位 Å
1.385
备注
指定cav时,除非solvent指定为water会自动使用默认值,其他溶剂必须手动指定 solventRho、 solventRadius。
指定rep或dis时,除非solvent指定为water会自动使用默认值,其他溶剂必须手动指定 solventRho、 solventAtoms。
一些常见溶剂的分子半径
Solvent |
Water |
Tetrahydrofuran |
Cyclohexane |
Methanol |
Ethanol |
Tetrachloromethane |
Radius(Å) |
1.385 |
2.900 |
2.815 |
1.855 |
2.180 |
2.685 |
计算色散排斥能以及孔穴能时,默认使用的Bondi半径,也可以自定义计算色散排斥能或者孔穴能时的半径。
通过 solventAtomicSASRadii 关键词来指定计算色散排斥能时所构建的SAS孔穴的溶剂分子中每类原子的半径。
通过 radiiForCavEnergy 关键词来指定计算孔穴能时的半径,并且可以通过 acidHRadiusForCavEnergy 关键词来单独设置酸性H的半径。
solventAtomicSASRadii # 计算色散排斥能时,构建SAS孔穴的溶剂分子中每类原子的半径
H=1.20 O=1.50
radiiForCavEnergy # 计算孔穴能的溶质半径
H=1.4430 O=1.7500 # 注意事项同radii
acidHRadiusForCavEnergy # 计算孔穴能的溶质半径,单独设置酸性H,单位 Å
1.2
非平衡溶剂化新理论简介
激发态溶剂化效应需要考虑非平衡溶剂化现象。溶剂的极化可以分为快极化和慢极化部分。垂直吸收和发射过程十分迅速,溶剂的偶极和构型不能迅速调整至与溶质电荷达到平衡的状态,于是需要考虑非平衡溶剂化效应。
传统非平衡溶剂化理论中平衡态到非平衡态的可逆功积分方式存在与热力学第二定律相矛盾的问题,会导致溶剂重组能的高估。在进行态特定计算时,采用了李象远教授课题组发展的非平衡溶剂化新理论(X. Y. Li. Int. J. Quantum Chem. 2015, 115(11): 700-721)。
激发态溶剂化效应计算
激发态溶剂化效应在隐式模型中有 线性响应 (linear-response, LR)和 态特定 (state-specific, SS)的处理方式。
垂直吸收计算
以下是采用 线性响应 计算甲醛分子激发态非平衡溶剂化效应的输入文件:
$COMPASS
Title
ch2o Molecule test run
Basis
6-31g
Geometry
C 0.00000000 0.00000000 -0.54200000
O 0.00000000 0.00000000 0.67700000
H 0.00000000 0.93500000 -1.08200000
H 0.00000000 -0.9350000 -1.08200000
END geometry
nosymm
unit
ang
$END
$xuanyuan
$END
$SCF
rks
dft
b3lyp
grid
medium
solvent
user # 用户指定
dielectric
78.3553 # 输入介电常数
opticalDielectric
1.7778 # 光介电常数
solmodel
iefpcm
$END
$TDDFT
iroot
8
solneqlr
$END
其中,在 TDDFT 中加入 solneqlr 关键词,表示要进行非平衡溶剂化效应计算。
备注
计算非平衡溶剂化效应时,溶剂如果为用户指定的(详见 BDF支持的溶剂类型列表 ),需要设置光介电常数,关键词为 opticalDielectric。
BDF目前支持一阶微扰态特定的能量计算(ptSS),以下是采用 态特定 计算丙烯醛分子激发态非平衡溶剂化效应的输入文件:
$COMPASS
Title
SS-PCM of S-trans-acrolein Molecule
Basis
cc-PVDZ
Geometry
C 0.55794100 -0.45384200 -0.00001300
H 0.44564200 -1.53846100 -0.00002900
C -0.66970500 0.34745600 -0.00001300
H -0.50375600 1.44863100 -0.00005100
C 1.75266800 0.14414300 0.00001100
H 2.68187400 -0.42304000 0.00001600
H 1.83151500 1.23273300 0.00002700
O -1.78758800 -0.11830000 0.00001600
END geometry
$END
$xuanyuan
$END
$SCF
rks
dft
PBE0
solvent
water
solmodel
iefpcm
$END
$TDDFT
iroot
5
istore
1
$END
$resp
nfiles
1
method
2
iroot
1 2 3
geom
norder
0
solneqss
$end
其中,在 resp 中加入 solneqss 关键词,表示要进行态特定非平衡溶剂化效应计算。指定 norder 为 0 ,表示不进行梯度的计算。用 iroot 指定计算哪些态。
在文件中的输出:
-Energy correction based on constrant equilibrium theory with relaxed density
*State 1 -> 0
Corrected vertical absorption energy = 3.6935 eV
Nonequilibrium solvation free energy = -0.0700 eV
Equilibrium solvation free energy = -0.1744 eV
其中 Corrected vertical absorption energy 表示采用李象远教授课题组发展的非平衡溶剂化新理论计算的激发能矫正。
上面的例子中,垂直吸收能为 \(3.69eV\)。
BDF目前还支持矫正的线性响应的计算(corrected linear response, cLR),以下是采用cLR计算丙烯醛分子激发态非平衡溶剂化效应的输入文件:
$COMPASS
Title
cLR-PCM of S-trans-acrolein Molecule
Basis
cc-PVDZ
Geometry
C 0.55794100 -0.45384200 -0.00001300
H 0.44564200 -1.53846100 -0.00002900
C -0.66970500 0.34745600 -0.00001300
H -0.50375600 1.44863100 -0.00005100
C 1.75266800 0.14414300 0.00001100
H 2.68187400 -0.42304000 0.00001600
H 1.83151500 1.23273300 0.00002700
O -1.78758800 -0.11830000 0.00001600
END geometry
$END
$xuanyuan
$END
$SCF
rks
dft
PBE0
solvent
water
solmodel
iefpcm
$END
$TDDFT
iroot
5
istore
1
$END
$TDDFT
iroot
5
istore
1
solneqlr
$END
$resp
nfiles
1
method
2
iroot
1
geom
norder
0
solneqlr
solneqss
$end
在文件中的找到 第一个TDDFT 的输出, 以及resp模块中的cLR输出:
No. 1 w= 3.7475 eV -191.566549 a.u. f= 0.0001 D<Pab>= 0.0000 Ova= 0.4683
CV(0): A( 15 )-> A( 16 ) c_i: 0.9871 Per: 97.4% IPA: 5.808 eV Oai: 0.4688
CV(0): A( 15 )-> A( 17 ) c_i: 0.1496 Per: 2.2% IPA: 9.144 eV Oai: 0.4392
...
Excitation energy correction(cLR) = -0.0377 eV
可算得cLR的激发能为 \(3.7475-0.0377=3.7098eV\)。
激发态几何优化
对于几何优化过程,溶剂有足够的时间进行响应,应考虑平衡溶剂效应。
需要在 tddft 以及 resp 模块中加入 soleqlr 关键词来表示平衡溶剂效应的计算。输入文件的其他部分以及输出,与 TDDFT相关章节 类似,此处不再赘述。
以下是苯酚分子的激发态溶剂化效应的几何优化计算
$COMPASS
Geometry
C -1.15617700 -1.20786100 0.00501300
C -1.85718200 0.00000000 0.01667700
C -1.15617700 1.20786100 0.00501300
C 0.23962700 1.21165300 -0.01258600
C 0.93461900 0.00000000 -0.01633400
C 0.23962700 -1.21165300 -0.01258600
H -1.69626800 -2.15127300 0.00745900
H -2.94368500 0.00000000 0.02907200
H -1.69626800 2.15127300 0.00745900
H 0.80143900 2.14104700 -0.03186000
H 0.80143900 -2.14104700 -0.03186000
O 2.32295900 0.00000000 -0.08796400
H 2.68364400 0.00000000 0.81225800
End geometry
basis
6-31G
$END
$bdfopt
solver
1
$end
$XUANYUAN
$END
$SCF
DFT
gb3lyp
rks
solModel
iefpcm
solvent
water
$END
$TDDFT
iroot
5
istore
1
soleqlr
$END
$resp
geom
soleqlr
method
2
nfiles
1
iroot
1
$end
垂直发射计算
在激发态的平衡几何结构下,进行ptSS或者cLR的平衡溶剂化效应的计算,将保存对应的溶剂慢极化电荷。在随后的scf模块中加入 emit 关键词,来计算非平衡的基态能量。以丙烯醛分子为例,采用ptSS计算激发态,对应的输入文件如下:
$COMPASS
Geometry
C -1.810472 0.158959 0.000002
H -1.949516 1.241815 0.000018
H -2.698562 -0.472615 -0.000042
C -0.549925 -0.413873 0.000029
H -0.443723 -1.502963 -0.000000
C 0.644085 0.314498 0.000060
H 0.618815 1.429158 -0.000047
O 1.862127 -0.113145 -0.000086
End geometry
basis
cc-PVDZ
$END
$XUANYUAN
$END
$SCF
DFT
PBE0
rks
solModel
iefpcm
solvent
water
$END
$TDDFT
iroot
5
istore
1
#soleqlr
$END
$resp
nfiles
1
method
2
iroot
1
geom
norder
0
#soleqlr
soleqss
$end
$SCF
DFT
PBE0
rks
solModel
iefpcm
solvent
water
emit
$END
需要注意指定 soleqss 来计算平衡溶剂化效应。在文件中的输出为:
-Energy correction based on constrant equilibrium theory
*State 1 -> 0
Corrected vertical emission energy = 2.8118 eV
Nonequilibrium solvation free energy = -0.0964 eV
Equilibrium solvation free energy = -0.1145 eV
其中 Corrected vertical emission energy 表示采用李象远教授课题组发展的非平衡溶剂化新理论计算的激发能矫正。
上面的例子中,垂直发射能为 \(2.81eV\)。
采用cLR计算时,需要在文件中的找到 第一个TDDFT 的输出, 以及resp模块中的cLR输出,并与两个scf的 E_tot 之差进行相加,得到最终的垂直发射能。
采用显式溶剂和隐式溶剂相结合的方法计算激发态溶剂化效应
激发态溶剂化效应可以采用显式溶剂和隐式溶剂相结合的方法计算。以水溶液为例,由于溶质分子的HOMO和LUMO轨道有可能弥散到 第一水合层,所以在进行激发态计算时可以将第一水合层的水分子包括在TDDFT计算区域,而其余部分用隐式溶剂处理。
以芥子酸(sinapic acid)为例。为了确定溶质分子的第一水合层,可以采用Amber程序将芥子酸分子置于小的水盒子中进行分子动力学模拟。 待体系平衡后,可分析溶质分子周围水分子分布情况,从而确定第一水合层。当然,也可以选取多帧结构进行计算,然后取平均。
水合层分子选取可以采用VMD程序完成。假设输入为pdb文件,在命令行中可以选择第一水合层分子,并保存为pdb文件。命令如下:
atomselect top "same resid as (within 3.5 of not water)" # 选择第一水合层
atomselect0 writepdb sa.pdb #溶质分子和第一水合层保存于pdb文件
上例中选取了与溶质分子相距3.5埃范围内的所有水分子,并且水分子的三个原子中只要有一个在截断范围内,就选择整个分子。选取结果如图所示:
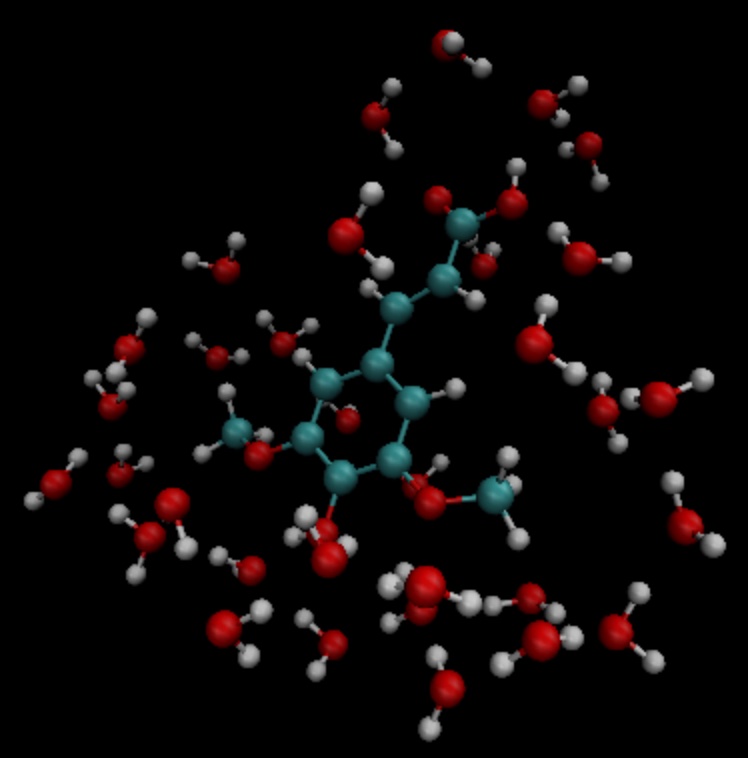
依据sa.pdb文件中的坐标信息,进行TDDFT计算,输入文件如下:
$COMPASS
Title
SA Molecule test run
Basis
6-31g
Geometry
C 14.983 14.539 6.274
C 14.515 14.183 7.629
C 13.251 14.233 8.118
C 12.774 13.868 9.480
C 11.429 14.087 9.838
C 10.961 13.725 11.118
O 9.666 13.973 11.525
C 8.553 14.050 10.621
C 11.836 13.125 12.041
O 11.364 12.722 13.262
C 13.184 12.919 11.700
O 14.021 12.342 12.636
C 15.284 11.744 12.293
C 13.648 13.297 10.427
O 14.270 14.853 5.341
O 16.307 14.468 6.130
H 15.310 13.847 8.286
H 12.474 14.613 7.454
H 10.754 14.550 9.127
H 7.627 14.202 11.188
H 8.673 14.888 9.924
H 8.457 13.118 10.054
H 10.366 12.712 13.206
H 15.725 11.272 13.177
H 15.144 10.973 11.525
H 15.985 12.500 11.922
H 14.687 13.129 10.174
H 16.438 14.756 5.181
O 18.736 9.803 12.472
H 18.779 10.597 11.888
H 19.417 10.074 13.139
O 18.022 14.021 8.274
H 17.547 14.250 7.452
H 18.614 13.310 7.941
O 8.888 16.439 7.042
H 9.682 16.973 6.797
H 8.217 17.162 7.048
O 4.019 14.176 11.140
H 4.032 13.572 10.360
H 4.752 14.783 10.885
O 16.970 8.986 14.331
H 17.578 9.273 13.606
H 17.497 8.225 14.676
O 8.133 17.541 10.454
H 8.419 17.716 11.386
H 8.936 17.880 9.990
O 8.639 12.198 13.660
H 7.777 11.857 13.323
H 8.413 13.155 13.731
O 13.766 11.972 4.742
H 13.858 12.934 4.618
H 13.712 11.679 3.799
O 10.264 16.103 14.305
H 9.444 15.605 14.054
H 10.527 15.554 15.084
O 13.269 16.802 3.701
H 13.513 16.077 4.325
H 14.141 17.264 3.657
O 13.286 14.138 14.908
H 13.185 14.974 14.393
H 13.003 13.492 14.228
O 16.694 11.449 15.608
H 15.780 11.262 15.969
H 16.838 10.579 15.161
O 7.858 14.828 14.050
H 7.208 15.473 13.691
H 7.322 14.462 14.795
O 15.961 17.544 3.706
H 16.342 16.631 3.627
H 16.502 17.866 4.462
O 10.940 14.245 16.302
H 10.828 13.277 16.477
H 11.870 14.226 15.967
O 12.686 10.250 14.079
H 11.731 10.151 14.318
H 12.629 11.070 13.541
O 9.429 11.239 8.483
H 8.927 10.817 7.750
H 9.237 12.182 8.295
O 17.151 15.141 3.699
H 17.124 14.305 3.168
H 18.133 15.245 3.766
O 17.065 10.633 9.634
H 16.918 10.557 8.674
H 17.024 9.698 9.909
O 17.536 14.457 10.874
H 18.014 13.627 11.089
H 17.683 14.460 9.890
O 5.836 16.609 13.299
H 4.877 16.500 13.549
H 5.760 16.376 12.342
O 19.014 12.008 10.822
H 18.249 11.634 10.308
H 19.749 11.655 10.256
O 15.861 14.137 15.750
H 14.900 13.990 15.574
H 16.185 13.214 15.645
O 11.084 9.639 10.009
H 11.641 9.480 9.213
H 10.452 10.296 9.627
O 14.234 10.787 16.235
H 13.668 10.623 15.444
H 13.663 10.376 16.925
O 14.488 8.506 13.105
H 13.870 9.136 13.550
H 15.301 8.683 13.628
O 14.899 17.658 9.746
H 15.674 18.005 9.236
H 15.210 16.754 9.926
O 8.725 13.791 7.422
H 9.237 13.488 6.631
H 8.845 14.770 7.309
O 10.084 10.156 14.803
H 9.498 10.821 14.366
H 10.215 10.613 15.669
O 5.806 16.161 10.582
H 5.389 16.831 9.993
H 6.747 16.470 10.509
O 6.028 13.931 7.206
H 5.971 14.900 7.257
H 6.999 13.804 7.336
O 17.072 12.787 2.438
H 16.281 12.594 1.885
H 17.062 11.978 3.013
END geometry
nosymm
mpec+cosx
$END
$xuanyuan
$end
$SCF
rks
dft
b3lyp
solvent
water
grid
medium
$END
# input for tddft
$tddft
iroot # Calculate 1 root for each irrep. By default, 10 roots are calculated
1 # for each irrep
memjkop # maxium memeory for Coulomb and Exchange operator. 1024 MW (Mega Words)
1024
$end
BDF中支持的溶剂类型列表
Name |
Short Name |
\({\epsilon}\) |
\({\epsilon_{opt}}\) |
|---|---|---|---|
water |
H2O |
78.3553 |
1.7764 |
acetic acid |
ACETACID |
6.2528 |
1.8824 |
acetone |
ACETONE |
20.4930 |
1.8463 |
acetonitrile |
ACETNTRL |
35.6880 |
1.8069 |
acetophenone |
ACETPHEN |
17.4400 |
2.3630 |
aniline |
ANILINE |
6.8882 |
2.5163 |
anisole |
ANISOLE |
4.2247 |
2.3025 |
benzaldehyde |
BENZALDH |
18.2200 |
2.3910 |
benzene |
BENZENE |
2.2706 |
2.2533 |
benzonitrile |
BENZNTRL |
25.5920 |
2.3375 |
benzyl chloride |
BENZYLCL |
6.7175 |
2.3688 |
1-bromo-2-methylpropane |
BRISOBUT |
7.7792 |
2.0587 |
bromobenzene |
BRBENZEN |
5.3954 |
2.4327 |
bromoethane |
BRETHANE |
9.0100 |
2.0275 |
bromoform |
BROMFORM |
4.2488 |
2.5616 |
1-bromooctane |
BROCTANE |
5.0244 |
2.1095 |
1-bromopentane |
BRPENTAN |
6.2690 |
2.0872 |
2-bromopropane |
BRPROPA2 |
9.3610 |
2.0309 |
1-bromopropane |
BRPROPAN |
8.0496 |
2.0572 |
butanal |
BUTANAL |
13.4500 |
1.9163 |
butanoic acid |
BUTACID |
2.9931 |
1.9544 |
1-butanol |
BUTANOL |
17.3320 |
1.9580 |
2-butanol |
BUTANOL2 |
15.9440 |
1.9538 |
butanone |
BUTANONE |
18.2460 |
1.9011 |
butanonitrile |
BUTANTRL |
24.2910 |
1.9160 |
butyl acetate |
BUTILE |
4.9941 |
1.9435 |
butylamine |
NBA |
4.6178 |
1.9687 |
n-butylbenzene |
NBUTBENZ |
2.3600 |
2.2195 |
sec-butylbenzene |
SBUTBENZ |
2.3446 |
2.2186 |
tert-butylbenzene |
TBUTBENZ |
2.3447 |
2.2282 |
carbon disulfide |
CS2 |
2.6105 |
2.6631 |
carbon tetrachloride |
CARBNTET |
2.2280 |
2.1319 |
chlorobenzene |
CLBENZEN |
5.6968 |
2.3229 |
sec-butyl chloride |
SECBUTCL |
8.3930 |
1.9519 |
chloroform |
CHCL3 |
4.7113 |
2.0906 |
1-chlorohexane |
CLHEXANE |
5.9491 |
2.0161 |
1-chloropentane |
CLPENTAN |
6.5022 |
1.9957 |
1-chloropropane |
CLPROPAN |
8.3548 |
1.9263 |
o-chlorotoluene |
OCLTOLUE |
4.6331 |
2.3311 |
m-cresol |
M-CRESOL |
12.4400 |
2.3833 |
o-cresol |
O-CRESOL |
6.7600 |
2.3596 |
cyclohexane |
CYCHEXAN |
2.0165 |
2.0352 |
cyclohexanone |
CYCHEXON |
15.6190 |
2.1045 |
cyclopentane |
CYCPENTN |
1.9608 |
1.9782 |
cyclopentanol |
CYCPNTOL |
16.9890 |
2.1112 |
cyclopentanone |
CYCPNTON |
13.5800 |
2.0638 |
cis-decalin |
DECLNCIS |
2.2139 |
2.1934 |
trans-decalin |
DECLNTRA |
2.1781 |
2.1594 |
decalin (cis/trans mixture) |
DECLNMIX |
2.1960 |
2.1765 |
n-decane |
DECANE |
1.9846 |
1.9887 |
1-decanol |
DECANOL |
7.5305 |
2.0655 |
1,2-dibromoethane |
EDB12 |
4.9313 |
2.3676 |
dibromomethane |
DIBRMETN |
7.2273 |
2.3778 |
dibutyl ether |
BUTYLETH |
3.0473 |
1.9578 |
o-dichlorobenzene |
ODICLBNZ |
9.9949 |
2.4072 |
1,2-dichloroethane |
EDC12 |
10.1250 |
2.0874 |
cis-dichloroethylene |
C12DCE |
9.2000 |
2.0996 |
trans-dichloroethylene |
T12DCE |
2.1400 |
2.0892 |
dichloromethane |
DCM |
8.9300 |
2.0283 |
diethyl ether |
ETHER |
4.2400 |
1.8295 |
diethyl sulfide |
ET2S |
5.7230 |
2.0822 |
diethylamine |
DIETAMIN |
3.5766 |
1.9221 |
diiodomethane |
MI |
5.3200 |
3.0363 |
diisopropyl ether |
DIPE |
3.3800 |
1.8712 |
dimethyl disulfide |
DMDS |
9.6000 |
2.3375 |
dimethyl sulfoxide |
DMSO |
46.8260 |
2.0079 |
n,n-dimethylacetamide |
DMA |
37.7810 |
2.0678 |
cis-1,2-dimethylcyclohexane |
CISDMCHX |
2.0600 |
2.0621 |
n,n-dimethylformamide |
DMF |
37.2190 |
2.0463 |
2,4-dimethylpentane |
DMEPEN24 |
1.8939 |
1.9085 |
2,4-dimethylpyridine |
DMEPYR24 |
9.4176 |
2.2530 |
2,6-dimethylpyridine |
DMEPYR26 |
7.1735 |
2.2359 |
1,4-dioxane |
DIOXANE |
2.2099 |
2.0232 |
diphenyl ether |
PHOPH |
3.7300 |
2.4923 |
dipropylamine |
DPROAMIN |
2.9112 |
1.9740 |
n-dodecane |
DODECAN |
2.0060 |
2.0209 |
1,2-ethanediol |
MEG |
40.2450 |
2.0501 |
ethanethiol |
ETSH |
6.6670 |
2.0478 |
ethanol |
ETHANOL |
24.8520 |
1.8526 |
ethyl acetate |
ETOAC |
5.9867 |
1.8832 |
ethyl formate |
ETOME |
8.3310 |
1.8493 |
ethylbenzene |
EB |
2.4339 |
2.2377 |
ethylphenyl ether |
PHENETOL |
4.1797 |
2.2729 |
fluorobenzene |
C6H5F |
5.4200 |
2.1562 |
1-fluorooctane |
FOCTANE |
3.8900 |
1.9418 |
formamide |
FORMAMID |
108.9400 |
2.0944 |
formic acid |
FORMACID |
51.1000 |
1.8807 |
n-heptane |
HEPTANE |
1.9113 |
1.9260 |
1-heptanol |
HEPTANOL |
11.3210 |
2.0303 |
2-heptanone |
HEPTNON2 |
11.6580 |
1.9847 |
4-heptanone |
HEPTNON4 |
12.2570 |
1.9794 |
n-hexadecane |
HEXADECN |
2.0402 |
2.0578 |
n-hexane |
HEXANE |
1.8819 |
1.8904 |
hexanoic acid |
HEXNACID |
2.6000 |
2.0059 |
1-hexanol |
HEXANOL |
12.5100 |
2.0102 |
2-hexanone |
HEXANON2 |
14.1360 |
1.9620 |
1-hexene |
HEXENE |
2.0717 |
1.9146 |
1-hexyne |
HEXYNE |
2.6150 |
1.9569 |
iodobenzene |
C6H5I |
4.5470 |
2.6244 |
1-iodobutane |
IOBUTANE |
6.1730 |
2.2503 |
iodoethane |
C2H5I |
7.6177 |
2.2901 |
1-iodohexadecane |
IOHEXDEC |
3.5338 |
2.1922 |
iodomethane |
CH3I |
6.8650 |
2.3654 |
1-iodopentane |
IOPENTAN |
5.6973 |
2.2377 |
1-iodopropane |
IOPROPAN |
6.9626 |
2.2674 |
isopropylbenzene |
CUMENE |
2.3712 |
2.2246 |
p-isopropyltoluene |
P-CYMENE |
2.2322 |
2.2228 |
mesitylene |
MESITYLN |
2.2650 |
2.2482 |
methanol |
METHANOL |
32.6130 |
1.7657 |
2-methoxyethanol |
EGME |
17.2000 |
1.9667 |
methyl acetate |
MEACETAT |
6.8615 |
1.8534 |
methyl benzoate |
MEBNZATE |
6.7367 |
2.2995 |
methyl butanoate |
MEBUTATE |
5.5607 |
1.9260 |
methyl formate |
MEFORMAT |
8.8377 |
1.8045 |
4-methyl-2-pentanone |
MIBK |
12.8870 |
1.9494 |
methyl propanoate |
MEPROPYL |
6.0777 |
1.8975 |
2-methyl-1-propanol |
ISOBUTOL |
16.7770 |
1.9474 |
2-methyl-2-propanol |
TERBUTOL |
12.4700 |
1.9260 |
n-methylaniline |
NMEANILN |
5.9600 |
2.4599 |
methylcyclohexane |
MECYCHEX |
2.0240 |
2.0252 |
n-methylformamide (E/Z mixture) |
NMFMIXTR |
181.5600 |
2.0503 |
2-methylpentane |
ISOHEXAN |
1.8900 |
1.8810 |
2-methylpyridine |
MEPYRID2 |
9.9533 |
2.2371 |
3-methylpyridine |
MEPYRID3 |
11.6450 |
2.2620 |
4-methylpyridine |
MEPYRID4 |
11.9570 |
2.2611 |
nitrobenzene |
C6H5NO2 |
34.8090 |
2.4218 |
nitroethane |
C2H5NO2 |
28.2900 |
1.9368 |
nitromethane |
CH3NO2 |
36.5620 |
1.9091 |
1-nitropropane |
NTRPROP1 |
23.7300 |
1.9650 |
2-nitropropane |
NTRPROP2 |
25.6540 |
1.9444 |
o-nitrotoluene |
ONTRTOLU |
25.6690 |
2.3870 |
n-nonane |
NONANE |
1.9605 |
1.9751 |
1-nonanol |
NONANOL |
8.5991 |
2.0543 |
5-nonanone |
NONANONE |
10.6000 |
2.0150 |
n-octane |
OCTANE |
1.9406 |
1.9527 |
1-octanol |
OCTANOL |
9.8629 |
2.0435 |
2-octanone |
OCTANON2 |
9.4678 |
2.0025 |
n-pentadecane |
PENTDECN |
2.0333 |
2.0492 |
pentanal |
PENTANAL |
10.0000 |
1.9444 |
n-pentane |
NPENTANE |
1.8371 |
1.8428 |
pentanoic acid |
PENTACID |
2.6924 |
1.9839 |
1-pentanol |
PENTANOL |
15.1300 |
1.9884 |
2-pentanone |
PENTNON2 |
15.2000 |
1.9307 |
3-pentanone |
PENTNON3 |
16.7800 |
1.9388 |
1-pentene |
PENTENE |
1.9905 |
1.8810 |
E-2-pentene |
E2PENTEN |
2.0510 |
1.9025 |
pentyl acetate |
PENTACET |
4.7297 |
1.9664 |
pentylamine |
PENTAMIN |
4.2010 |
2.0967 |
perfluorobenzene |
PFB |
2.0290 |
1.8981 |
phenylmethanol |
BENZALCL |
12.4570 |
2.3704 |
propanal |
PROPANAL |
18.5000 |
1.8594 |
propanoic acid |
PROPACID |
3.4400 |
1.9235 |
1-propanol |
PROPANOL |
20.5240 |
1.9182 |
2-propanol |
PROPNOL2 |
19.2640 |
1.8978 |
propanonitrile |
PROPNTRL |
29.3240 |
1.8646 |
2-propen-1-ol |
PROPENOL |
19.0110 |
1.9980 |
propyl acetate |
PROPACET |
5.5205 |
1.9160 |
propylamine |
PROPAMIN |
4.9912 |
1.9238 |
pyridine |
PYRIDINE |
12.9780 |
2.2786 |
tetrachloroethene |
C2CL4 |
2.2680 |
2.2659 |
tetrahydrofuran |
THF |
7.4257 |
1.9740 |
tetrahydrothiophene-s,s-dioxide |
SULFOLAN |
43.9620 |
2.2002 |
tetralin |
TETRALIN |
2.7710 |
2.3756 |
thiophene |
THIOPHEN |
2.7270 |
2.3375 |
thiophenol |
PHSH |
4.2728 |
2.5259 |
toluene |
TOLUENE |
2.3741 |
2.2383 |
tributyl phosphate |
TBP |
8.1781 |
2.0232 |
1,1,1-trichloroethane |
TCA111 |
7.0826 |
2.0676 |
1,1,2-trichloroethane |
TCA112 |
7.1937 |
2.1650 |
trichloroethene |
TCE |
3.4220 |
2.1824 |
triethylamine |
ET3N |
2.3832 |
1.9628 |
2,2,2-trifluoroethanol |
TFE222 |
26.7260 |
1.6659 |
1,2,4-trimethylbenzene |
TMBEN124 |
2.3653 |
2.2644 |
2,2,4-trimethylpentane |
ISOCTANE |
1.9358 |
1.9363 |
n-undecane |
UNDECANE |
1.9910 |
2.0730 |
m-xylene |
M-XYLENE |
2.3478 |
2.2416 |
o-xylene |
O-XYLENE |
2.5454 |
2.2665 |
p-xylene |
P-XYLENE |
2.2705 |
2.2374 |
xylene (mixture) |
XYLENEMX |
2.3879 |
2.2485 |
1,1-dichloroethane |
10.1900 |
2.0880 |
|
1-iodopentene |
5.7800 |
2.2350 |
|
1-pentyne |
2.0600 |
1.9182 |
|
2-chlorobutane |
8.3900 |
1.9656 |
|
benzyl alcohol |
11.9200 |
2.3716 |
|
bromooctane |
5.0200 |
2.1083 |
|
butyl ethanoate |
5.0700 |
1.9432 |
|
butyl benzene |
2.3600 |
2.2201 |
|
carbon tet |
2.2300 |
2.1316 |
|
chlorotoluene |
6.8500 |
2.3654 |
|
decalin |
2.1900 |
2.1934 |
|
dimethylacetamide |
DMAC |
37.7800 |
2.0678 |
dimethylformamide |
DMF |
37.2200 |
2.0478 |
dimethylpyridine |
7.1700 |
2.2350 |
|
dodecane |
2.0100 |
2.0221 |
|
E-1,2-dichloroethene |
2.1400 |
2.0880 |
|
ethyl ethanoate |
6.0800 |
1.8824 |
|
ethyl methanoate |
8.3300 |
1.8469 |
|
ethyl eneglycol |
40.2500 |
2.0506 |
|
hexadecyl iodide |
3.5300 |
2.1934 |
|
hexanoic |
2.6000 |
2.0051 |
|
isobutanol |
16.7800 |
1.9460 |
|
isopropyl ether |
3.8800 |
1.8714 |
|
isopropyl toluene |
2.2300 |
2.2231 |
|
methyl ethanoate |
6.8600 |
1.8523 |
|
methyl methanoate |
8.8400 |
1.8036 |
|
methyl phenyl ketone |
17.4400 |
2.3624 |
|
methylformamide |
181.5600 |
2.0506 |
|
hexadecane |
2.0600 |
2.0592 |
|
methylaniline |
5.9600 |
2.4649 |
|
pentane |
1.8400 |
1.8414 |
|
pentadecane |
2.0300 |
2.0478 |
|
pentyl ethanoate |
4.7300 |
1.9656 |
|
phenyl ether |
3.7300 |
2.4932 |
|
propyl ethanoate |
5.5200 |
1.9155 |
|
pyrrolidine |
8.0400 |
2.0822 |
|
sec-butanol |
15.9400 |
1.9544 |
|
t-butanol |
12.4700 |
1.9238 |
|
t-butylbenzene |
2.3400 |
2.2290 |
|
tetrahyrothiophenedioxide |
43.9600 |
2.1993 |
|
tribromomethane |
4.2500 |
2.5632 |
|
trichloromethane |
TCM |
4.7100 |
2.0909 |
Z-1,2-dichloroethene |
9.2000 |
2.0996 |
|
isoquinoline |
11.0000 |
1.0100 |
|
quinoline |
9.1600 |
1.0100 |
|
diethylether |
4.2400 |
1.8295 |
|
dichloroethane |
10.1250 |
2.0874 |
|
carbontetrachloride |
2.2280 |
2.1319 |
|
heptane |
1.9113 |
1.9260 |
|
dimethylsulfoxide |
46.8260 |
2.0079 |
|
argon |
1.4300 |
1.4300 |
|
krypton |
1.5190 |
1.5190 |
|
xenon |
1.7060 |
1.7060 |
|
n-octanol |
9.8629 |
2.0435 |
|
aceticacid |
6.2528 |
1.8824 |
|
a-chlorotoluene |
6.7175 |
2.3688 |
|
benzylalcohol |
12.4570 |
2.3704 |
|
butanoicacid |
2.9931 |
1.9544 |
|
butylethanoate |
4.9941 |
1.9435 |
|
carbondisulfide |
2.6105 |
2.6631 |
|
decalin-mixture |
2.1960 |
2.1106 |
|
dibutylether |
3.0473 |
1.9578 |
|
diethylsulfide |
5.7230 |
2.0822 |
|
diisopropylether |
3.3800 |
1.8712 |
|
dimethyldisulfide |
9.6000 |
2.3375 |
|
diphenylether |
3.7300 |
2.4923 |
|
E-1,2-dichloroethene |
2.1400 |
2.0892 |
|
E-2-pentene |
2.0510 |
1.9025 |
|
ethylethanoate |
5.9867 |
1.8832 |
|
ethylmethanoate |
8.3310 |
1.8493 |
|
ethylphenylether |
4.1797 |
2.2729 |
|
formicacid |
51.1000 |
1.8807 |
|
hexanoicacid |
2.6000 |
2.0059 |
|
methylbenzoate |
6.7367 |
2.2995 |
|
methylbutanoate |
5.5607 |
1.9260 |
|
methylethanoate |
6.8615 |
1.8534 |
|
methylmethanoate |
8.8377 |
1.8045 |
|
methylpropanoate |
6.0777 |
1.8975 |
|
n-methylaniline |
5.9600 |
2.4599 |
|
n-methylformamide-mixture |
181.5600 |
2.0503 |
|
n,n-dimethylacetamide |
37.7810 |
2.0678 |
|
n,n-dimethylformamide |
37.2190 |
2.0463 |
|
pentanoicacid |
2.6924 |
1.9839 |
|
pentylethanoate |
4.7297 |
1.9664 |
|
propanoicacid |
3.4400 |
1.9235 |
|
propylethanoate |
5.5205 |
1.9160 |
|
tetrahydrothiophene-s,s-dioxide |
43.9620 |
2.2002 |
|
tributylphosphate |
8.1781 |
2.0232 |
|
xylene-mixture |
2.3879 |
2.2485 |
|
Z-1,2-dichloroethene |
9.2000 |
2.0996 |